
|
|||||||||||||||||||||||||||||||||||||||||
|
News Archive
Data sets of the year (2013/12/31)
Technical, Biological and Clinical.  This week we are highlighting the three finest examples of proteomics data made public in 2013. As we
have been doing since 2010, we are naming the best data in three categories. These ratings
do not take into account the associated publication: only the data itself was considered in these
awards. Any of these data sets would be ideal for use as standards in the development of
computational biology methods associated with proteomics.
This week we are highlighting the three finest examples of proteomics data made public in 2013. As we
have been doing since 2010, we are naming the best data in three categories. These ratings
do not take into account the associated publication: only the data itself was considered in these
awards. Any of these data sets would be ideal for use as standards in the development of
computational biology methods associated with proteomics.
Data set of the week: (2013/12/22)
Proteome and phosphoproteome characterization reveals new response and defense mechanisms of Brachypodium distachyon leaves under salt stress. Overall rating: ") very good data (specialist interest) very good data (specialist interest)
 This data set consisted of 87 results,
corresponding to either large LC/MS/MS runs of phosphopeptide enriched samples or small sets of spectra from individually excised 2D gel electrophoresis bands.
The data files were made available through ProteomeXchange, PXD000340 (LC/MS/MS) and
PXD000470 (2D).
It has been published by
Lv DW, Subburaj S, Cao M, Yan X, Li X, Appels R, Sun DF, Ma W and Yan YM,
Mol Cell Proteomics. 2013 Dec 11 (PubMed).
This data set consisted of 87 results,
corresponding to either large LC/MS/MS runs of phosphopeptide enriched samples or small sets of spectra from individually excised 2D gel electrophoresis bands.
The data files were made available through ProteomeXchange, PXD000340 (LC/MS/MS) and
PXD000470 (2D).
It has been published by
Lv DW, Subburaj S, Cao M, Yan X, Li X, Appels R, Sun DF, Ma W and Yan YM,
Mol Cell Proteomics. 2013 Dec 11 (PubMed).
This study highlights the utility of the model species
Brachypodium distachyon (purple false brome),
which has the potential to be used as a model for economically important temperate grasses, e.g. Triticum spp., in
the same way that A. thaliana is used as a model for the economically important Brassicaceae species. Its genome and
proteome have been sequenced and its relatively simple genetics (compared to the hexaploid wheat) make it a much more
tractible platform for research. The data presented here were produced by well done experiments. It is also one of few
publicly available studies that uses MALDI TOF/TOF to identify excised 2D gel bands. This type of data has a very different
character than that generated by the more commonly used shotgun proteomics methods, making it interesting from an bioinformatics
point-of-view. Rather than trying to unravel a complex mixture of peptides from all of the proteins in a sample, this type of
data has a small number of spectra (typically ≤ 10) that correspond to a single protein species (splice variant/post-translationally
modified sequence). Anyone interested in emphasizing the consequences of individual identifications should look carefully at this
data, which may have considerable utility for the construction of tutorial material.
Data set of the week: (2013/12/15)
Global analysis of Cdc14 dephosphorylation sites reveals essential regulatory role in mitosis and cytokinesis. Overall rating: very good data (specialist interest)
 This data set consisted of 9 results,
corresponding to LC/MS/MS analyses of samples enriched in phosphopeptides using IMAC purification.
The data files were made available through ProteomeXchange, PXD000375.
It has been published by
Kao L, Wang YT, Chen YC, Tseng SF, Jhang JC, Chen YJ and Teng SC,
Mol Cell Proteomics. 2013 Dec 7 (PubMed).
This data set consisted of 9 results,
corresponding to LC/MS/MS analyses of samples enriched in phosphopeptides using IMAC purification.
The data files were made available through ProteomeXchange, PXD000375.
It has been published by
Kao L, Wang YT, Chen YC, Tseng SF, Jhang JC, Chen YJ and Teng SC,
Mol Cell Proteomics. 2013 Dec 7 (PubMed).
The results of this study demonstrate that IMAC sample preparation methods can be
quite reproducible at both the protein and peptide levels. The biological question being examined was the converse
of many studies that involve IMAC: rather than looking for kinase-induced protein phosphorylation, the study was designed to find
phosphorylase-induced protein dephosphorylation events. The intepretation of the study was made more complicated by apparent HPLC
timing problems in setting the start of mass spectrum data acquistion and the termination of the previous gradient.
Data set of the week: (2013/12/8)
U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer's disease. Overall rating: very good data (specialist interest)
 This data set consisted of 80 results,
generated from LC/MS/MS analyses of SDS-PAGE gel bands.
The data files were made available through ProteomeXchange, PXD000067.
It has been published by
Bai B, Hales CM, Chen PC, Gozal Y, Dammer EB, Fritz JJ, Wang X, Xia Q, Duong DM, Street C, Cantero G, Cheng D, Jones DR, Wu Z, Li Y, Diner I, Heilman CJ, Rees HD, Wu H, Lin L, Szulwach KE, Gearing M, Mufson EJ, Bennett DA, Montine TJ, Seyfried NT, Wingo TS, Sun YE, Jin P, Hanfelt J, Willcock DM, Levey A, Lah JJ and Peng J,
Proc Natl Acad Sci U S A. 2013 Oct 8;110(41):16562-7 (PubMed).
This data set consisted of 80 results,
generated from LC/MS/MS analyses of SDS-PAGE gel bands.
The data files were made available through ProteomeXchange, PXD000067.
It has been published by
Bai B, Hales CM, Chen PC, Gozal Y, Dammer EB, Fritz JJ, Wang X, Xia Q, Duong DM, Street C, Cantero G, Cheng D, Jones DR, Wu Z, Li Y, Diner I, Heilman CJ, Rees HD, Wu H, Lin L, Szulwach KE, Gearing M, Mufson EJ, Bennett DA, Montine TJ, Seyfried NT, Wingo TS, Sun YE, Jin P, Hanfelt J, Willcock DM, Levey A, Lah JJ and Peng J,
Proc Natl Acad Sci U S A. 2013 Oct 8;110(41):16562-7 (PubMed).
This study attempted to characterize the proteins present in the insoluble aggregates obtained from
the brains of Alzheimer's Disease patients. The results demonstrated that a surprisingly large number of proteins were detectable
in these aggregates, including many cytoskeletal, mitochondrial and ribonuclear proteins. The results were of sufficient quality that they can serve
as a guide to the proteins expected in this type of preparation.
Data set of the week: (2013/12/1)
Identification of telomere-associated molecules by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP). Overall rating: very good data (specialist interest)
 This data set consisted of 30 results,
generated from affinity pull-down experiments.
The data files were made available through ProteomeXchange, PXD000461.
It has been published by
Fujita T, Asano Y, Ohtsuka J, Takada Y, Saito K, Ohki R and Fujii H
Sci Rep. 2013 Nov 8;3:3171 (PubMed).
This data set consisted of 30 results,
generated from affinity pull-down experiments.
The data files were made available through ProteomeXchange, PXD000461.
It has been published by
Fujita T, Asano Y, Ohtsuka J, Takada Y, Saito K, Ohki R and Fujii H
Sci Rep. 2013 Nov 8;3:3171 (PubMed).
This paper reports a method for obtaining proteins specifically enriched in those physically associated with a particular chromosomal
feature, namely the telomere. The results suggests that the method works as designed: the resulting lists of proteins show
significant enrichment in chromatin associated proteins, nuclear ribonucleoproteins as well as a number of relatively rarely
observed proteins, such as Rhox5:p and Runx1t1:p. Some of the chromatograms show an odd dropout of signal in the latter half of
the run. This puzzling feature does not diminish the potential utility of the method being demonstrated: it should encourage
other groups to reproduce and improve upon the results reported.
Data set of the week: (2013/11/24)
Shotgun proteomics suggests involvement of additional enzymes in dioxin degradation by Sphingomonas wittichii RW1. Overall rating: very good data (specialist interest)
 This data set consisted of 84 results,
generated from LC/MS/MS experiments.
The data files were made available through ProteomeXchange, PXD000403.
It has been published by
Hartmann EM and Armengaud J
Environ Microbiol. 2013 Aug 30 (PubMed).
This data set consisted of 84 results,
generated from LC/MS/MS experiments.
The data files were made available through ProteomeXchange, PXD000403.
It has been published by
Hartmann EM and Armengaud J
Environ Microbiol. 2013 Aug 30 (PubMed).
Sphingomonas wittichii is an environmental bacterium that can effectively metabolize
toxic pollutants such as dioxin into benign products. This study provides a nice insight into the proteins expressed by
this organism in response to changes in the presence of high concentrations of pollutant molecules in the
growth medium. The experiments were very consistently performed, with highly reproducible chromatographic and mass spectrometric
methods applied.
Data set of the week: (2013/11/17)
Rhesus Cytomegalovirus pp65 limits viral dissemination but is dispensable for persistence and immunity. Overall rating: very good data (specialist interest)
 This data set consisted of 8 results,
generated from LC/MS/MS experiments performed on infected Macaca mulatta cells.
The data files were made available through PeptideAtlas, PASS00367.
It has been submitted for publication by
Malouli D, Hansen SG, Nakayasu ES, Marshall EE, Hughes CM, Ventura AB, Gilbride RM, Lewis MS, Xu G, Kreklywich C, Whizin NO, Fischer MB, Legasse AW, Viswanathan K, Siess DC, Camp D, Axthelm MK, Kahl C, DeFilippis VR, Smith RD, Streblow DN, Picker LJ, and Frueh K.
This data set consisted of 8 results,
generated from LC/MS/MS experiments performed on infected Macaca mulatta cells.
The data files were made available through PeptideAtlas, PASS00367.
It has been submitted for publication by
Malouli D, Hansen SG, Nakayasu ES, Marshall EE, Hughes CM, Ventura AB, Gilbride RM, Lewis MS, Xu G, Kreklywich C, Whizin NO, Fischer MB, Legasse AW, Viswanathan K, Siess DC, Camp D, Axthelm MK, Kahl C, DeFilippis VR, Smith RD, Streblow DN, Picker LJ, and Frueh K.
The results of this study were unusual, as they are dominated by viral proteins from the infectious agent,
Macacine herpesvirus 3. Herpes viruses have unusually large proteomes compared to many other types of viruses: this
particular virus has 225 proteins. Many of the viral proteins were observed in most eight analyses, giving anyone interested in this
virus a good starting point for comparison with their own experimental findings. The general applicability of the results was
somewhat reduced by the extent of peptide carbamylation caused by the sample prepartion protocol.
GPMDB equipment outage (2013/11/18)
 For several hours today (between about 18:00 and 21:00 UTC) the main host computer for
GPMDB was behaving erratically, because of an equipment problem. The problem seems to
be repaired, but we will continue to monitor the system's behavior for the next few
hours to be sure that the fix worked properly. Sorry for any inconvenience.
For several hours today (between about 18:00 and 21:00 UTC) the main host computer for
GPMDB was behaving erratically, because of an equipment problem. The problem seems to
be repaired, but we will continue to monitor the system's behavior for the next few
hours to be sure that the fix worked properly. Sorry for any inconvenience.
Data set of the week: (2013/11/10)
Host-centric Proteomics of Stool: A Novel Strategy Focused on intestinal Responses to the Gut Microbiota. Overall rating: ") excellent data (worth study) excellent data (worth study)
 This data set consisted of 265 results,
generated from LC/MS/MS experiments performed on mouse stool.
The data files were made available through ProteomeXchange, PXD000240.
It was published by
Lichtman JS, Marcobal A, Sonnenburg JL, and Elias JE in
Mol Cell Proteomics. 2013 Nov;12(11):3310-8 (PubMed).
This data set consisted of 265 results,
generated from LC/MS/MS experiments performed on mouse stool.
The data files were made available through ProteomeXchange, PXD000240.
It was published by
Lichtman JS, Marcobal A, Sonnenburg JL, and Elias JE in
Mol Cell Proteomics. 2013 Nov;12(11):3310-8 (PubMed).
This data set provides an excellent example of how to study a normally difficult subject: the interaction
of the gut microbiome with the host-secreted gut proteome. The study uses bacterially-naive mice and populates their
gut with a specific set of common human intestinal microbes to provide a model of the human digestive process. Both the
proteins secreted by the mouse and those of the known prokaryotes can be easily detected, even though the original sample
is less than ideal for proteomics. Any one examining the data should keep in mind that the samples contain high concentrations
of host organism proteases and peptidases, resulting in numerous non-tryptic peptides that must be treated properly to obtain
a good idea of the proteins present.
Data set of the week: (2013/11/2)
The One Hour Yeast Proteome. Overall rating: very good data (specialist interest)
 This data set consisted of 7 results,
generated from LC/MS/MS experiments performed on yeast lysates.
The data files were made available through the Chorus Project.
It was published by
Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS and Coon JJ in
Mol Cell Proteomics. 2013 Oct 19 (PubMed).
This data set consisted of 7 results,
generated from LC/MS/MS experiments performed on yeast lysates.
The data files were made available through the Chorus Project.
It was published by
Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS and Coon JJ in
Mol Cell Proteomics. 2013 Oct 19 (PubMed).
This paper is one of a spate of recent publications demonstrating the capabilities
of new experimental methods and instruments by analyzing cell lysates of lab-grown S. cerevisiae (including the L-A and L-BC viruses). In this
case, the study utilizes a new mass spectrometer that is capable of taking twice as many spectra per hour as
the previous generation of instrument. The results demonstrated that the additional spectra generated could
be used as efficiently as the older instrument configuration, resulting in good quality peptide sequence
assignments of as many as 65% of the spectra. The group make unusually good use of the chromatographic run, only
losing ~10% of the run to isocratic holds and column washes. The only puzzling feature of the data was the
parent ion mass accuracy: the new instrument's accuracy appears to be ± 8 ppm FWHM, compared to
the older instrument's ± 2 ppm FWHM.
Data set of the week: (2013/10/24)
Phosphoproteomic analysis implicates the mTORC2-FoxO1 axis in VEGF signaling and feedback activation of receptor tyrosine kinases. Overall rating: ") excellent data (leading the field) excellent data (leading the field)
 This data set consisted of 7 results,
generated from LC/MS/MS experiments performed on samples after phospho-peptide enrichment.
The data files were made available through Massive, MSV000078325.
It was published by
Zhuang G, Yu K, Jiang Z, Chung A, Yao J, Ha C, Toy K, Soriano R, Haley B, Blackwood E, Sampath D, Bais C, Lill JR and Ferrara N in
Sci Signal. 2013 Apr 16;6(271):ra25 (PubMed).
This data set consisted of 7 results,
generated from LC/MS/MS experiments performed on samples after phospho-peptide enrichment.
The data files were made available through Massive, MSV000078325.
It was published by
Zhuang G, Yu K, Jiang Z, Chung A, Yao J, Ha C, Toy K, Soriano R, Haley B, Blackwood E, Sampath D, Bais C, Lill JR and Ferrara N in
Sci Signal. 2013 Apr 16;6(271):ra25 (PubMed).
This study provides some straightforward insights into the complexities of VEGF signalling. The experiments
used iTRAQ-based quantitation and high resolution mass spectrometry to generate clean, unambiguous identifications with unusually
high overall statistical significance. Any group interested in dissecting signalling involving serine-threonine or tyrosine kinases
should carefully consider the methods used by the study.
Data set of the week: (2013/10/15)
Proteomic investigation of the time course responses of RAW 264.7 macrophages to infection with Salmonella enterica. Overall rating: excellent data (worth study)
 This data set consisted of 29 results,
generated from LC/MS/MS experiments performed on whole cell preparations.
The data files were made available through ProteomeXchange, PXD000389.
It was published by
Shi L, Chowdhury SM, Smallwood HS, Yoon H, Mottaz-Brewer HM, Norbeck AD, McDermott JE, Clauss TR, Heffron F, Smith RD and Adkins JN in
Infect Immun. 2009 77:3227-33 (PubMed).
This data set consisted of 29 results,
generated from LC/MS/MS experiments performed on whole cell preparations.
The data files were made available through ProteomeXchange, PXD000389.
It was published by
Shi L, Chowdhury SM, Smallwood HS, Yoon H, Mottaz-Brewer HM, Norbeck AD, McDermott JE, Clauss TR, Heffron F, Smith RD and Adkins JN in
Infect Immun. 2009 77:3227-33 (PubMed).
The data associated with this study has recently been made publicly available and it provides an interesting insight into
the proteins from prokaryotes that can be detected in infected eukaryote cells. In these
experiments, the time course of Salmonella entrica typhimurium intracellular infection in transformed mouse macrophages was explored. Both the mouse cell proteome's response
to infection and the gradual emergence of the S. enterica proteome in the cells can be tracked. Any group interested in the proteomics
of infectious disease or the detection of microbiome proteomes in intact organisms would benefit from examining the results. The study also
provides some very good examples of the gag:p, gag-pol:p and env:p proteins from the Moloney murine leukemia virus, one of the
retroviruses necessary for the initial transformation of the mouse cell line.
Data set of the week: (2013/10/5)
Quantitative proteomic analysis of amniocytes reveals potentially dysregulated molecular networks in Down syndrome. Overall rating: very good data (specialist interest)
 This data set consisted of 30 results,
generated from a combination of multidimensional chromatography and SILAC quantitation.
The data files were made available through ProteomeXchange, PXD000385.
It was published by
Cho CK, Drabovich AP, Karagiannis GS, Martínez-Morillo E, Dason S, Dimitromanolakis A, Diamandis EP in
Clin Proteomics. 2013 10:2 (PubMed).
This data set consisted of 30 results,
generated from a combination of multidimensional chromatography and SILAC quantitation.
The data files were made available through ProteomeXchange, PXD000385.
It was published by
Cho CK, Drabovich AP, Karagiannis GS, Martínez-Morillo E, Dason S, Dimitromanolakis A, Diamandis EP in
Clin Proteomics. 2013 10:2 (PubMed).
This study was designed to determine the quantitative characteristics of the detectable proteome of
amniocytes (cells floating freely in amniotic fluid) obtained from patients with Down Syndrome. The experiments
were well executed and the mass spectrometry very consistently performed. The chromatographic method used resulted in unusually
good recovery of hydrophobic (late eluting) peptides. The method also seemed somewhat biased against hydrophilic
(early eluting) peptides, which should be kept in mind by any group reproducing these measurements.
Data set of the week: (2013/9/29)
In-depth proteomic analysis of mouse microglia using a combination of FASP and StageTip-based, high pH, reversed-phase fractionation. Overall rating: excellent data (worth study)
 This data set consisted of 36 results,
using a novel strategy for peptide fractionation prior to LC/MS/MS.
The data files were made available through ProteomeXchange, PXD000168.
It was published by
Han D, Moon S, Kim Y, Kim J, Jin J and Kim Y in
Proteomics 2013 Aug 14 (PubMed).
This data set consisted of 36 results,
using a novel strategy for peptide fractionation prior to LC/MS/MS.
The data files were made available through ProteomeXchange, PXD000168.
It was published by
Han D, Moon S, Kim Y, Kim J, Jin J and Kim Y in
Proteomics 2013 Aug 14 (PubMed).
These studies contain the best information to date on the proteome of the mouse BV-2 cell
line, which was originally derived from microglial cells. The sample preparation methods used performed very
well, producing excellent data and continuing a recent trend of protocols that lead to the recovery of phosphorylated peptides without
any specific enrichment protocol. The results also showed very few experimental artifacts,
reproducible chromatography and good run-to-run consistency in the high resolution MS/MS parent and fragment ion calibrations.
The 3516th daily build of GPMDB (Sept, 20, 2013) contained 1,002,098,817 peptide identifications, marking the
first time GPMDB exceeded one billion entries. We would like to thank the proteomics community for making
so much data obtained from so many species pubicly available, giving us the opportunity to display the results
to the biomedical and scientific research communities.
Data set of the week: (2013/9/20)
SILAC-based proteomics of human primary endothelial cell morphogenesis unveils tumor angiogenic markers. Overall rating: ") very good data (general interest) very good data (general interest)
 This data set consisted of 452 results,
consisting of LC-MS/MS runs of individual gel bands.
The data files were made available through ProteomeXchange, PXD000359.
It was published by
Zanivan S, Maione F, Hein MY, Hernández-Fernaud JR, Ostasiewicz P, Giraudo E and Mann M in
Mol Cell Proteomics 2013 Aug 26 (PubMed).
This data set consisted of 452 results,
consisting of LC-MS/MS runs of individual gel bands.
The data files were made available through ProteomeXchange, PXD000359.
It was published by
Zanivan S, Maione F, Hein MY, Hernández-Fernaud JR, Ostasiewicz P, Giraudo E and Mann M in
Mol Cell Proteomics 2013 Aug 26 (PubMed).
While multidimensional peptide separation technology is a very frequently used method in proteomics,
much of the biomedical research community still depends on (and trusts) SDS-PAGE gel separation as their
standard method of protein sample preparation. The experiments described in this manuscript show how well proteomics
can perform when using gel-based separations and SILAC quantitation methods to solve a genuine biological problem. This
relatively large dataset was well constructed and the associated mass spectrometry data was very consistent from run-to-run.
Any group interested in the proteomics of endothelial cells should consult these results to get an idea of
the proteins accessible using this type of experiment.
Data set of the week: (2013/9/11)
Proteomic analysis of the SLP76 interactome in resting and activated primary mast cells. Overall rating: excellent data (worth study)
 This data set consisted of 32 results,
from bait pull-down affinity purification experiments.
The data files were made available through ProteomeXchange, PXD000052.
It was published by
Bounab Y, Hesse AM, Iannascoli B, Grieco L, Coute Y, Niarakis A, Roncagalli R, Lie E, Lam KP, Demangel C, Thieffry D, Garin J, Malissen B and Daeron M in
Mol Cell Proteomics 2013 Jul 2 (PubMed).
This data set consisted of 32 results,
from bait pull-down affinity purification experiments.
The data files were made available through ProteomeXchange, PXD000052.
It was published by
Bounab Y, Hesse AM, Iannascoli B, Grieco L, Coute Y, Niarakis A, Roncagalli R, Lie E, Lam KP, Demangel C, Thieffry D, Garin J, Malissen B and Daeron M in
Mol Cell Proteomics 2013 Jul 2 (PubMed).
This set of experiments does an excellent job of demonstrating how complex affinity pull-down experiments have become because
of the improved sensitivity of proteomics techniques and instrumentation. The results were very clean, with a minimum of experimental
artifacts and a well-reproduced chromatographic protocol. The data also showed surprisingly good recovery of phosphopeptides, which are often
lost in studies of this type unless special care is taken.
Data set of the week: (2013/9/6)
Quantitative proteomic analysis of microdissected breast cancer tissues: comparison of label-free and SILAC based quantification with shotgun, directed and targeted MS approaches. Overall rating: very good data (general interest)
 This data set consisted of 57 results,
from 1D chromatographic separations coupled to LC/MS/MS.
The data files were made available through ProteomeXchange, PXD000278.
It was published by
Liu NQ, Dekker LJ, Stingl C, Güzel C, De Marchi T, Martens JW, Foekens JA, Luider TM and Umar A. in
J Proteome Res. 2013 Aug 20 (PubMed).
This data set consisted of 57 results,
from 1D chromatographic separations coupled to LC/MS/MS.
The data files were made available through ProteomeXchange, PXD000278.
It was published by
Liu NQ, Dekker LJ, Stingl C, Güzel C, De Marchi T, Martens JW, Foekens JA, Luider TM and Umar A. in
J Proteome Res. 2013 Aug 20 (PubMed).
This study does a very good job of demonstrating which proteins could be readily identified and
quantified from a straightforward laser microdissection protocol used on real tissue samples. Overall, the samples were
worked up consistently and the chromatography was consistently well done. The data showed that the proteins were not
chemically degraded by the laser microdissection method used and that it was a viable tissue preparation technique
for proteomics studies.
We have compiled a comprehensive list of all human protein
ubiquitination sites represented by good quality data in GPMDB. This list has been subdivided on a chromosome-by-chromosome
basis, using ENSEMBL v. 70 as the source of the protein and gene sequences. All of the splice variants listed
by ENSEMBL have been annotated.
The files associated with the annotation for each chromosome (and a merged list of all chromosomes) is now available
by FTP. A description of the
format of these files
(README.txt) is
in the same directory. A short summary of the number of ubiquitin-modified proteins, genes and sites is given
here.
For unique protein sequences in the proteome, the overall totals are as follows:
Data set of the week: (2013/8/27)
Global Proteome Analysis of the NCI-60 Cell Line Panel. Overall rating: very good data (general interest)
 This data set consisted of 899 results,
from experiments aimed at either broad surveys of the full proteome or kinome of all 59 cell lines and high sensitivity analysis of 9 selected cell lines.
The data files were made available through Proteomics DB.
It was published by
Moghaddas Gholami A, Hahne H, Wu Z, Auer FJ, Meng C, Wilhelm M and Kuster B in
Cell Rep. 2013 4:609-20 (PubMed).
This data set consisted of 899 results,
from experiments aimed at either broad surveys of the full proteome or kinome of all 59 cell lines and high sensitivity analysis of 9 selected cell lines.
The data files were made available through Proteomics DB.
It was published by
Moghaddas Gholami A, Hahne H, Wu Z, Auer FJ, Meng C, Wilhelm M and Kuster B in
Cell Rep. 2013 4:609-20 (PubMed).
This study did a good job of generating a large, heterogenous (but quite consistent) data set from a variety of standard
Homo sapiens cell lines that have been widely used in biomedical research. Any researcher interested in a source of good quality peptide MS/MS data
for almost any purpose should consider this data set carefully. The range of workflows used and the types of experiments performed were representative
on current proteomics technology, with a strong emphasis on reproducibility and standardization of techniques.
Data set of the week: (2013/8/15)
Phosphoproteomic analyses reveal extensive rewiring of the host phosphoprotein network during gammaherpesvirus replication. Overall rating: very good data (specialist interest)
 This data set consisted of 12 results,
from experiments using a phosphopeptide affinity enrichment workflow.
The data files were made available through Chorus.
It was published by
Stahl JA, Chavan SS, J Sifford , Macleod V, Voth D, Edmondson RD and Forrest JC. in
Plos Pathogens 2013 (in press).
This data set consisted of 12 results,
from experiments using a phosphopeptide affinity enrichment workflow.
The data files were made available through Chorus.
It was published by
Stahl JA, Chavan SS, J Sifford , Macleod V, Voth D, Edmondson RD and Forrest JC. in
Plos Pathogens 2013 (in press).
The information made available in this study is the first public data set that does a good job of sketching out simultaneously the protein phosphorylation
patterns in both an infectious agent (in this case murid herpesvirus 4) and the host (Mus musculus). The extent of phosphorylation of the viral proteins
was significant and reproducible within the study. Hopefully this type of experiment will be performed more frequently, given the clinical importance of
herpesvirus infections in mammalian hosts.
Data set of the week: (2013/8/6)
Comparison of detergent-based sample preparation workflows for LTQ-Orbitrap analysis of the Escherichia coli proteome. Overall rating: excellent data (worth study)
 This data set consisted of 131 results,
from a variety of different laboratory workflows.
The data files were made available through PeptideAtlas, PASS00111.
It was published by
Tanca A, Biosa G, Pagnozzi D, Addis MF and Uzzau S in
Proteomics 2013 Jun 20 (PubMed).
This data set consisted of 131 results,
from a variety of different laboratory workflows.
The data files were made available through PeptideAtlas, PASS00111.
It was published by
Tanca A, Biosa G, Pagnozzi D, Addis MF and Uzzau S in
Proteomics 2013 Jun 20 (PubMed).
This study does a good job of providing information that can be used to understand the effects of specific experimental
workflows on the outcomes of proteomics analysis. By starting with the same sample and using multiple protein extraction/fractionation methods,
the resulting data sets show the details of a protein's detection can vary both quantitatively and qualitatively depending on how the sample
was treated. These results should be
very useful both for planning experiments and for general tutorial purposes.
Data set of the week: (2013/8/1)
Comparative phosphoproteomic analysis of checkpoint recovery identifies new regulators of the DNA damage response. Overall rating: very good data (general interest)
 This data set consisted of 247 results,
generated by multidimensional chromatography and titanium dioxide affinity purification using isotopically labelled dimethylation for relative quantitation.
The data files were made available through ProteomeXchange, PXD000222.
It was published by
Halim VA, Alvarez-Fernández M, Xu YJ, Aprelia M, van den Toorn HW, Heck AJ, Mohammed S and Medema RH. in
Sci Signal. 2013 Apr 23;6(272):rs9 (PubMed).
This data set consisted of 247 results,
generated by multidimensional chromatography and titanium dioxide affinity purification using isotopically labelled dimethylation for relative quantitation.
The data files were made available through ProteomeXchange, PXD000222.
It was published by
Halim VA, Alvarez-Fernández M, Xu YJ, Aprelia M, van den Toorn HW, Heck AJ, Mohammed S and Medema RH. in
Sci Signal. 2013 Apr 23;6(272):rs9 (PubMed).
These results provide an interesting insight into the use of a simple isotope-labelled chemical tag (N-dimethyl)
as a quantitation reagent in the pursuit of a relatively rare sub-class of peptides, in this case phospho-peptides. The experiments were
uniformly well done, although some artifacts (such as carbamylation) could be detected throughout. Any one interested in the analysis
of data generated using this reagent for quantitation should consider these results as a very good set of examples
for understanding the LOD/LOQs possible in state-of-the-art measurements.
GPMDB adds new peptide-to-spectrum assignments incrementally once daily, resulting in a new "build" (today's build is #3464).
We track the number of new assignments at the peptide, protein and residue levels and make these historical numbers available by
FTP. We use this information to track the health of the system
and to understand seasonal fluctuations in the availability of new data.
A combination of factors has led to significant increase in the number of new peptide-to-spectrum assignments this year. Faster, higher
accuracy instruments, an increased emphasis on making raw data available and the availability of easy-to-use raw data repositories have
all contributed to this burst of new assignments. The histogram below shows the number of new peptide-to-spectrum assignments
added in 30 day increments for the period February-July 2013.
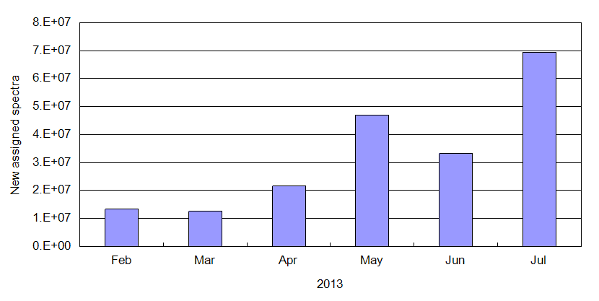
Data set of the week: (2013/7/25)
Adaptation of a commonly used, chemically defined medium for human embryonic stem cells to stable isotope labeling with amino acids in cell culture. Overall rating: very good data (specialist interest)
 This data set consisted of 13 results,
generated by LC/MS from human embryonic stem cell cultures, with lysine/arginine SILAC for quantitation.
The data files were made available through ProteomeXchange, PXD000151.
It was published by
Liberski AR, Al-Noubi MN, Rahman ZH, Halabi NM, Dib SS, Al-Mismar R, Billing AM, Krishnankutty R, Ahmad FS, Raynaud CM, Rafii A, Engholm-Keller K and Graumann J in
J Proteome Res. 2013 Jul 5;12:3233-45 (PubMed).
This data set consisted of 13 results,
generated by LC/MS from human embryonic stem cell cultures, with lysine/arginine SILAC for quantitation.
The data files were made available through ProteomeXchange, PXD000151.
It was published by
Liberski AR, Al-Noubi MN, Rahman ZH, Halabi NM, Dib SS, Al-Mismar R, Billing AM, Krishnankutty R, Ahmad FS, Raynaud CM, Rafii A, Engholm-Keller K and Graumann J in
J Proteome Res. 2013 Jul 5;12:3233-45 (PubMed).
The work focusses mainly on demonstrating that SILAC quantitation can be adapted to human embryonic stem cell cultures
that use "feeder-free" methods (i.e., cell culture without a substrate layer of mouse embryonic fibroblasts). The data clearly
supported the
stated goal of the study, but it also provides a very good resource for anyone requiring good SILAC data to demonstrate the utility of an algorithm
or analysis method. The results were very clean, with few contaminants and a minimum of experimental artifacts. The chromatography was well done
and consistent from run-to-run.
Data set of the week: (2013/7/15)
In-depth Proteomic Analysis of Mouse Cochlear Sensory Epithelium by Mass Spectrometry. Overall rating: very good data (specialist interest)
 This data set consisted of 160 results,
generated by LC/MS from samples prepared using several different protein and peptide fractionation methods.
The data files were made available through ProteomeXchange, PXD000231.
It was published by
Darville LN and Sokolowski BH in
J Proteome Res. 2013 Jun 26 (PubMed).
This data set consisted of 160 results,
generated by LC/MS from samples prepared using several different protein and peptide fractionation methods.
The data files were made available through ProteomeXchange, PXD000231.
It was published by
Darville LN and Sokolowski BH in
J Proteome Res. 2013 Jun 26 (PubMed).
This study exemplifies why the isolation of unusual tissues is so important in the proteomics
of organisms. The investigators isolated protein fractions from murine cochlear sensory epithelium tissue and performed
a series of protein and peptide fractionation studies in an effort to find low concentration proteins in this very specialized
tissue. The results show an unusually large number of previously unobserved proteins, as well as many of the best observations
of previously found, but rare, extracellular matrix proteins.
Data set of the week: (2013/7/6)
Proteomic characterization of Pseudomonas aeruginosa PAO1 inner membrane. Overall rating: excellent data (worth study)
 This data set consisted of 6 results,
each one a single LC-MS/MS analysis of a membrane preparation.
The data files were made available through ProteomeXchange, PXD000107.
It was published by
Casabona MG, Vandenbrouck Y, Attree I and Couté Y in
Proteomics 2013 Jun 7 (PubMed).
This data set consisted of 6 results,
each one a single LC-MS/MS analysis of a membrane preparation.
The data files were made available through ProteomeXchange, PXD000107.
It was published by
Casabona MG, Vandenbrouck Y, Attree I and Couté Y in
Proteomics 2013 Jun 7 (PubMed).
While it is a commonplace statement in proteomics that "membrane" proteins
are difficult to detect, this group has done an admirable job generating six very reproducible
data sets of the inner membrane proteome associated with the common environmental prokaryote Pseudomonas aeruginosa.
The data is top-notch, with an excellent combination of experimental workflow and chromatography that clearly
demonstrates that membrane proteins can be readily observed when sufficient care is taken in their preparation.
Data set of the week: (2013/6/27)
The Escherichia coli Phosphotyrosine Proteome Relates to Core Pathways and Virulence. Overall rating: very good data (general interest)
 This data set consisted of 24 results, each an LC-MS/MS analysis of
a pulldown affinity purification experiment using an anti-phosphotyrosine antibody.
The data files were made available through MASSIVE, MSV000077310.
It was published by
Hansen AM, Chaerkady R, Sharma J, Díaz-Mejía JJ, Tyagi N, Renuse S, Jacob HK, Pinto SM, Sahasrabuddhe NA, Kim MS, Delanghe B, Srinivasan N, Emili A, Kaper JB and Pandey A in
PLoS Pathog. 2013 Jun;9(6):e1003403 (PubMed).
This data set consisted of 24 results, each an LC-MS/MS analysis of
a pulldown affinity purification experiment using an anti-phosphotyrosine antibody.
The data files were made available through MASSIVE, MSV000077310.
It was published by
Hansen AM, Chaerkady R, Sharma J, Díaz-Mejía JJ, Tyagi N, Renuse S, Jacob HK, Pinto SM, Sahasrabuddhe NA, Kim MS, Delanghe B, Srinivasan N, Emili A, Kaper JB and Pandey A in
PLoS Pathog. 2013 Jun;9(6):e1003403 (PubMed).
While the existence of tyrosine kinase activity in Escherichia coli has been known for some time, this data represents
the first solid confirmation of protein tyrosine phosphorylation in this clinically important prokaryote. The data also provides evidence
for the range of proteins exhibiting tyrosine phosphorylation in two strains, both the commonly used lab strain E. coli K12 str. MG1655 as well
as the pathogenic strain E. coli O157:H7 str. EDL933. The level of phosphotyrosine enrichment is somewhat less than would be normally
found in similar eukaryote-based experiments, but the signals very clearly demonstrate the existence of this modification in this species.
Data set of the week: (2013/6/20)
The functional interactome landscape of the human histone deacetylase family. Overall rating: excellent data (worth study)
 This data set consisted of 75 results comprised mainly of
affinity pull-down experiments using conventional LC-MS/MS.
The data files were made available through ProteomeXchange, PXD000208.
It was published by
Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI and Cristea IM in
Mol Syst Biol. 2013 Jun 11;9:672 (PubMed).
This data set consisted of 75 results comprised mainly of
affinity pull-down experiments using conventional LC-MS/MS.
The data files were made available through ProteomeXchange, PXD000208.
It was published by
Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI and Cristea IM in
Mol Syst Biol. 2013 Jun 11;9:672 (PubMed).
This study provides clear insight into the benefits of affinity purification in proteomics. Many of the proteins
represented in this data set are the best observations of those protein species to date. The measurements are well-done, with
very consistent chromatography and mass spectrometry techniques employed. These spectra should be of interest to anyone interested in the
post-translational modifications associated with HDAC proteins.
As part of our contribution to the Human Proteome Project, we have compiled a comprehensive list of all human protein
lysine acetylation sites represented by good quality data in GPMDB. This list has been subdivided on a chromosome-by-chromosome
basis, using ENSEMBL v. 70 as the source of the protein and gene sequences. All of the splice variants listed
by ENSEMBL have been annotated.
The files associated with the annotation for each chromosome (and a merged list of all chromosomes) is now available
by FTP. A description of the
format of these files
(README.txt) is
in the same directory. A short summary of the number of lysine-acetylated proteins, genes and sites is given
here.
For unique protein sequences in the proteome, the overall totals are as follows:
As part of our contribution to the Human Proteome Project, we have compiled a comprehensive list of all human protein
N-terminal acetylation sites represented by good quality data in GPMDB. This list has been subdivided on a chromosome-by-chromosome
basis, using ENSEMBL v. 70 as the source of the protein and gene sequences. All of the splice variants listed
by ENSEMBL have been annotated. Protein N-terminal acetylation is the result of a multienzyme system
that is responsible for processing nascent polypeptide chains as they emerge from the ribosome (see Thomas Arnesen's
interesting review for details).
The files associated with the annotation for each chromosome (and a merged list of all chromosomes) is now available
by FTP. A description of the
format of these files
(README.txt) is
in the same directory. A short summary of the number of NT-acetylated proteins, genes and sites is given
here.
For unique protein sequences in the proteome, the overall totals are as follows:
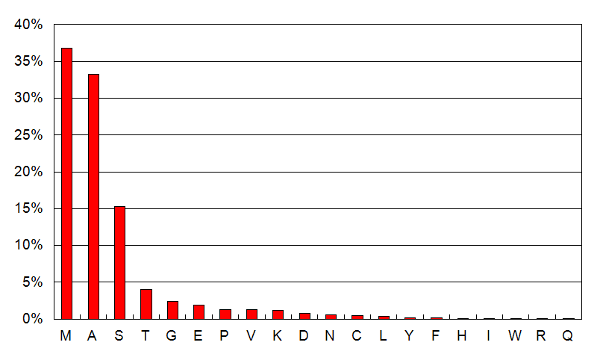 The fraction of residues found with NT-acetylation as a function of residue type, for all chromosomes (spreadsheet). 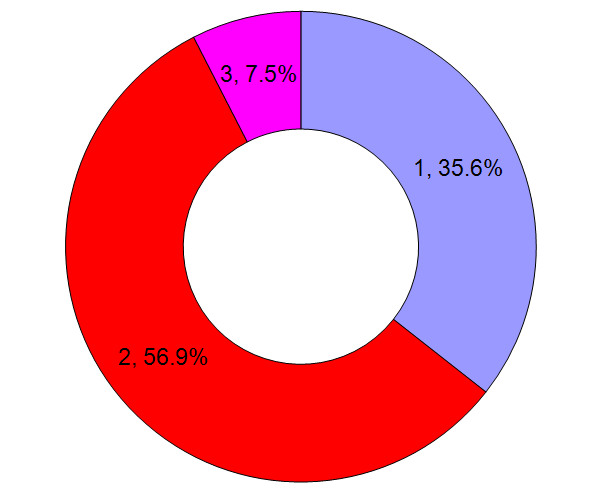 The observed position of the mature protein N-terminus and the corresponding fraction of all N-terminal acetylation, for all chromosomes (spreadsheet). Data set of the week: (2013/6/7)
Phosphoproteome dynamics reveal novel ERK1/2 MAP kinase substrates with broad spectrum of functions. Overall rating: very good data (general interest)
 This data set consisted of 124 results comprised mainly of
metal-oxide affinity purified samples run using conventional LC-MS/MS.
The data files were made available through PeptideAtlas, PASS00138.
It was published by
Courcelles M, Frémin C, Voisin L, Lemieux S, Meloche S, and Thibault P in
Mol Syst Biol. 2013 May 28;9:669 (PubMed).
This data set consisted of 124 results comprised mainly of
metal-oxide affinity purified samples run using conventional LC-MS/MS.
The data files were made available through PeptideAtlas, PASS00138.
It was published by
Courcelles M, Frémin C, Voisin L, Lemieux S, Meloche S, and Thibault P in
Mol Syst Biol. 2013 May 28;9:669 (PubMed).
This well-done study is the source of a significant amount of new data associated about Rattus norvegicus phosphopeptides.
Even though the rat is a common model species, there have been few phosphorylation studies using rat cell lines in which the raw data has been
made publicly available compared to the large volume of human and mouse studies that have been made public. The data provided here was very good quality and
should provide a useful starting point for any group interested in comparing the patterns of phosphopeptide detection across mammalian model species.
Data set of the week: (2013/6/1)
Variation and genetic control of protein abundance in humans. Overall rating: excellent data (leading the field)
 This data set consisted of 51 results comprised of
multidimensional chromatography run summaries.
The data files were made available through PeptideAtlas, PASS00230.
It was published by
Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, Tang H and Snyder M. in
Nature. 2013 May 15 (PubMed).
This data set consisted of 51 results comprised of
multidimensional chromatography run summaries.
The data files were made available through PeptideAtlas, PASS00230.
It was published by
Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, Tang H and Snyder M. in
Nature. 2013 May 15 (PubMed).
One of the milestone works
on lymphocyte protomics has been the 2007 work Lingfen Wu, et al., "Global survey of human T leukemic cells by integrating proteomics and transcriptomics profiling"
(PubMed). This new work (also with Lingfen Wu as a lead author) combines state-of-the-art RNASeq measurements with
very consistent deep proteomics data from cell lines derived by human herpesvirus 4
transformation of lymphoblasts from the individuals used in the HapMap project. This study has enough data
to make a good run at understanding the relationship between RNA and protein detectability using some of
the best instrumentation for detecting both types of macromolecule.
Data set of the week: (2013/5/24)
Modified MuDPIT Separation Identified 4488 Proteins in a System-wide Analysis of Quiescence in Yeast. Overall rating: very good data (specialist interest)
 This data set consisted of 135 results comprised of
multidimensional chromatography runs and experiment summaries.
The data files were made available through the the authors' web site.
It was published by
Webb KJ, Xu T, Park SK and Yates JR 3rd in
J Proteome Res. 2013 12:2177-84 (PubMed).
This data set consisted of 135 results comprised of
multidimensional chromatography runs and experiment summaries.
The data files were made available through the the authors' web site.
It was published by
Webb KJ, Xu T, Park SK and Yates JR 3rd in
J Proteome Res. 2013 12:2177-84 (PubMed).
This study provides a good insight into the proteins, peptides and post-translational modifications
that can be readily accessed in S. cerevisiae and its associated viruses (L-A and L-AB) under several different physiological conditions
using two different multidimensional chromatography protocols. The data was of good quality and nicely demonstrated the effects of the temporal details of
chromatographic conditions on spectrum identifications rates. The results also indicated the degree to which phosphorylations can now be detected in routine
analysis.
Data set of the week: (2013/5/16)
Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq. Overall rating: excellent data (leading the field)
 This data set consisted of 28 results that comprised a single
multidimensional chromatography experiment.
The data files were made available through PASSEL (PASS00215).
It was published by
Sheynkman GM, Shortreed MR, Frey BL and Smith LM in
Mol Cell Proteomics 2013 Apr 29 (PubMed).
This data set consisted of 28 results that comprised a single
multidimensional chromatography experiment.
The data files were made available through PASSEL (PASS00215).
It was published by
Sheynkman GM, Shortreed MR, Frey BL and Smith LM in
Mol Cell Proteomics 2013 Apr 29 (PubMed).
This data set defines the state-of-the-art with respect to "deep" proteomics
of a human cell line (Jurkat cells). The combination of a first dimension using high pH HPLC followed by
low pH HPLC produced a very well separated collection of peptides. The use of HCD coupled with high resolution
fragment ion measurements using an Orbitrap lead to very high confidence peptide assignments. Anyone interested in
detecting relatively rare post-translational modifications or determining splice variants would be well served
by performing their analysis on this data set first.
In addition to the human phosphorylation annotation released on Sunday, we have also prepared annotation in the
same format for a set of model species commonly used in proteomics experiments. Annotation for the following
species is now available:
C. elegans,
D. melanogaster,
M. musculus and
S. cerevisiae.
As part of our contribution to the Human Proteome Project, we have compiled a comprehensive list of all human protein
phosphorylation sites represented by good quality data in GPMDB. This list has been subdivided on a chromosome-by-chromosome
basis, using ENSEMBL v. 70 as the source of the protein and gene sequences. All of the splice variants listed
by ENSEMBL have been annotated.
The files associated with the annotation for each chromosome (and a merged list of all chromosomes) is now available
by FTP. A description of the
format of these files
(README.txt) is
in the same directory. A short summary of the number of phospho-proteins, genes and sites is given
here.
For unique protein sequences in the proteome, the overall totals are as follows:
Data set of the week: (2013/5/10)
Proteogenomic Analysis of Human Colon Carcinoma Cell Lines LIM1215, LIM1899, and LIM2405. Overall rating: very good data (specialist interest)
 This data set consisted of 136 results composed of individual SDS-PAGE gel slices
and experiment summaries.
The data files were made available through ProteomeXchange (PXD000120).
It was published by
Fanayan S, Smith JT, Lee LY, Yan F, Snyder M, Hancock WS and Nice E in
J Proteome Res. 2013 Mar 13 (PubMed).
This data set consisted of 136 results composed of individual SDS-PAGE gel slices
and experiment summaries.
The data files were made available through ProteomeXchange (PXD000120).
It was published by
Fanayan S, Smith JT, Lee LY, Yan F, Snyder M, Hancock WS and Nice E in
J Proteome Res. 2013 Mar 13 (PubMed).
The data reported here was a good example of what can be done with whole cell lysates
analyzed using SDS-PAGE protein separations and low resolution (LTQ) mass spectrometry. The experiments elucidate
an interesting biological issue: "How different were the protein concentrations in three related cell lines and how
were those changes generated by differences in RNA concentration?" This data would be useful for anyone interested
in practical difficulties associated with combining protein molecular mass information with peptide identifications
when using SDS-PAGE gels for protein separations.
 As some readers may have noticed, the logo for GPM and GPMDB has changed recently (thanks to noted electronic artist KD Thornton).
This change is part of a general redesign of the site to conform to more modern web page coding trends, simplify page navigation and improve
the usefulness of the overall site on smaller screens and mobile platforms. If you have any suggestions regarding things you would
like to see in a new design (or things that really bug you about the current one), please let us know at contact@thegpm.org.
The definition of the GPMDB REST interface has been expanded to include a new method, allowing the rapid
calculation of peptide ω frequencies for any set of peptides and protein accession numbers
stored in GPMDB. These frequencies are useful when comparing observed peptides to those previously observed:
a technical definition is given here.
The description of this method and an example have been added to the GPMDB Wiki page
for the REST interface.
As some readers may have noticed, the logo for GPM and GPMDB has changed recently (thanks to noted electronic artist KD Thornton).
This change is part of a general redesign of the site to conform to more modern web page coding trends, simplify page navigation and improve
the usefulness of the overall site on smaller screens and mobile platforms. If you have any suggestions regarding things you would
like to see in a new design (or things that really bug you about the current one), please let us know at contact@thegpm.org.
The definition of the GPMDB REST interface has been expanded to include a new method, allowing the rapid
calculation of peptide ω frequencies for any set of peptides and protein accession numbers
stored in GPMDB. These frequencies are useful when comparing observed peptides to those previously observed:
a technical definition is given here.
The description of this method and an example have been added to the GPMDB Wiki page
for the REST interface.
 Links to the "Protein Abundance Across Organisms" project (PaxDB)
have been added to the protein-specific display pages in GPM and GPMDB for appropriate species. These links can
be found by clicking the green "Protein" links button at the top of the appropriate pages. The PaxDB
system (Wang, M. et al. Mol Cell Proteomics 2012, doi:10.1074/mcp.O111.014704)
is, in the words of the developers:
Links to the "Protein Abundance Across Organisms" project (PaxDB)
have been added to the protein-specific display pages in GPM and GPMDB for appropriate species. These links can
be found by clicking the green "Protein" links button at the top of the appropriate pages. The PaxDB
system (Wang, M. et al. Mol Cell Proteomics 2012, doi:10.1074/mcp.O111.014704)
is, in the words of the developers:
PaxDB is a comprehensive absolute protein abundance database, which contains whole genome protein abundance
information across organisms and tissues. In PaxDB, the publicly available experimental data are imported
and mapped onto a common namespace and, in the case of tandem mass spectrometry data, re-processed using our
in-house standardized spectral counting pipeline.
Data set of the week: (2013/5/3)
Cell type-specific nuclear pores: a case in point for context-dependent stoichiometry of molecular machines. Overall rating: excellent data (leading the field)
 This data set consisted of 30 results,
each representing a sub-cellular fractionation experiment analyzed by reversed-phase HPLC.
The data files were made available through PASSEL (PASS00190).
It was published by
Ori A, Banterle N, Iskar M, Andrés-Pons A, Escher C, Khanh Bui H, Sparks L, Solis-Mezarino V, Rinner O, Bork P, Lemke EA, and Beck M in
Mol Syst Biol. 2013 Mar 19; 9:648 (PubMed).
This data set consisted of 30 results,
each representing a sub-cellular fractionation experiment analyzed by reversed-phase HPLC.
The data files were made available through PASSEL (PASS00190).
It was published by
Ori A, Banterle N, Iskar M, Andrés-Pons A, Escher C, Khanh Bui H, Sparks L, Solis-Mezarino V, Rinner O, Bork P, Lemke EA, and Beck M in
Mol Syst Biol. 2013 Mar 19; 9:648 (PubMed).
This study represents a thorough examination of the proteins associated with an important intracellular structure,
the human nuclear pore. A variety of methods were applied to the problem, such as protein chemistry isolations, proteomics, high resolution optical and electron microscopies, and the study tries to synthesize all of these measurements together to obtain a model of how nuclear pores differ between common cell lines (SK-MEL5, RKO, HEK 293 and HeLa). The proteomics data was excellent and demonstrates how well these techniques can be used as a component in the examination of a complex problem.
Data set of the week: (2013/4/25)
The Protein Interaction Landscape of the Human CMGC Kinase Group. Overall rating: excellent data (worth study)
 This data set consisted of 114 results,
each representing an affinity-purification experiment analyzed by reversed-phase HPLC.
The data files were made available through PASSEL (PASS00226).
It was published by
Varjosalo M, Keskitalo S, Van Drogen A, Nurkkala H, Vichalkovski A, Aebersold R and Gstaiger M in
Cell Reports 2013 25 April, 3(4):1306–20 (PubMed).
This data set consisted of 114 results,
each representing an affinity-purification experiment analyzed by reversed-phase HPLC.
The data files were made available through PASSEL (PASS00226).
It was published by
Varjosalo M, Keskitalo S, Van Drogen A, Nurkkala H, Vichalkovski A, Aebersold R and Gstaiger M in
Cell Reports 2013 25 April, 3(4):1306–20 (PubMed).
The data presented with this manuscript will give any interested investigator significant insight into the work
necessary to turn a set of identifications into a high-quality protein-protein interaction map. The proteomics experiments were consistently
done, with attention to the often overlooked chromatographic details. Anyone interested in performing or analyzing this type of information
would be well served by examining these experiments prior to planning their own. One feature of the data not commented on in the manuscript
were the clear signals for Human adenovirus C
E1A and E1B proteins (these proteins are coded on the viral DNA responsible for the original
HEK 293 transformation). These proteins are commonly observed in HEK 293 proteomics data, but the presence of these
proteins in specific pull-down experiments here may shed some light on their role in CMGC kinase signalling, which to our knowledge has not yet
been examined.
Data set of the week: (2013/4/18)
A Chemical Proteomics Approach to Profiling the ATP-Binding Proteome of Mycobacterium tuberculosis. Overall rating: very good data (specialist interest)
 This data set consisted of 33 results,
each representing chemical modifications, pull-downs and reversed-phase HPLC peptide separations.
The data files were made available through ProteomeXchange (PXD000141).
It was published by
Wolfe LM, Veeraraghavan U, Idicula-Thomas S, Schurer S, Wennerberg K, Reynolds R, Besra GS, and Dobos KM in
Mol Cell Proteomics. 2013 Mar 5 (PubMed).
This data set consisted of 33 results,
each representing chemical modifications, pull-downs and reversed-phase HPLC peptide separations.
The data files were made available through ProteomeXchange (PXD000141).
It was published by
Wolfe LM, Veeraraghavan U, Idicula-Thomas S, Schurer S, Wennerberg K, Reynolds R, Besra GS, and Dobos KM in
Mol Cell Proteomics. 2013 Mar 5 (PubMed).
This data represents a particularly interesting class of proteomics experiments: the combination of specific chemical probes
that facilitate protein and peptide purification techniques that very specifically target particular pathways and/or biochemistry. In this case,
it is the incorperation of desthiobiotin into proteins that bind ATP. This type of experiment does not require cutting-edge high throughput instruments,
rather its value is associated with developing a coherent model of how well the modification reagent is functioning in the experimental system
and then evaluating the results in the context of that model and the known biochemistry of the system.
Data set of the week: (2013/4/11)
The human leukocyte antigen-presented ligandome of B lymphocytes. Overall rating: very good data (specialist interest)
 This data set consisted of 191 results,
each representing a combination of peptide separation methods and reversed-phase HPLC peptide separations.
The data files were made available through PASSEL (PASS00211).
It was published by
Hassan C, Kester MG, Ru AH, Hombrink P, Drijfhout JW, Nijveen H, Leunissen JA, Heemskerk MH, Falkenburg JH, and Veelen PA in
Mol Cell Proteomics. 2013 Mar 19 (PubMed).
This data set consisted of 191 results,
each representing a combination of peptide separation methods and reversed-phase HPLC peptide separations.
The data files were made available through PASSEL (PASS00211).
It was published by
Hassan C, Kester MG, Ru AH, Hombrink P, Drijfhout JW, Nijveen H, Leunissen JA, Heemskerk MH, Falkenburg JH, and Veelen PA in
Mol Cell Proteomics. 2013 Mar 19 (PubMed).
This study was an interesting investigation of how to purify and detect a special class of endogenous peptides: those presented on the
surface of leukocytes by human leukocyte antigen class I molecules. These peptides are relatively short (~9 residues) and are generated by
ubiquitin-mediated proteolysis in the cell's proteasome. The peptides are also present at relatively low concentrations in normal cells and since they
have no special properties, they can be difficult to isolate. The protocols described in this paper do a good job of purifying these peptides and
demonstrated the merits of different peptide separations methods when applied to this situation. Any group interested in the large-scale analysis of
HLA class I peptides should examine the data in this work carefully.
Data set of the week: (2013/4/4)
Extensive Mass Spectrometry-Based Analysis of the Fission Yeast Proteome: The S. pombe PeptideAtlas. Overall rating: very good data (general interest)
 This data set consisted of 384 results,
each representing a combination of protein separation methods and reversed-phase HPLC peptide separations.
The data files were made available through PASSEL (PASS00069).
It was published by
Gunaratne J, Schmidt A, Quandt A, Neo SP, Sarac OS, Gracia T, Loguercio S, Ahrne E, Li Hai Xia R, Tan KH, Loessner C, Bahler J, Beyer A, Blackstock W, and Aebersold R in
Mol Cell Proteomics. 2013 Mar 5 (PubMed).
This data set consisted of 384 results,
each representing a combination of protein separation methods and reversed-phase HPLC peptide separations.
The data files were made available through PASSEL (PASS00069).
It was published by
Gunaratne J, Schmidt A, Quandt A, Neo SP, Sarac OS, Gracia T, Loguercio S, Ahrne E, Li Hai Xia R, Tan KH, Loessner C, Bahler J, Beyer A, Blackstock W, and Aebersold R in
Mol Cell Proteomics. 2013 Mar 5 (PubMed).
The experiments described in this paper were an attempt to characterize the proteome of the yeast Schizosaccharomyces pombe using
modern instruments and standard sample preparation methods. The laboratory model organism S. pombe is descended from an environmental species and
it has been extensively studied for determining the mechanisms behind its cell cycle and genetics. S. pombe has been less studied by proteomics methods
than its other beer-related relative, S. cerevisiae, and this study does a lot to address that deficit.
 Yesterday (April 2, 2013) marked the tenth anniversary of the first public release of the proteomics
search engine X! Tandem. At the time, it was the first open source protein identification software project to become available (a previous
academic effort had been attempted but was shutdown because of patent concerns). The release of the X! Tandem code – and the subsequent release a few months later
of the NCBI-sponsored open source search engine OMSSA – ushered in the modern era of proteomics informatics and computational biology.
Yesterday (April 2, 2013) marked the tenth anniversary of the first public release of the proteomics
search engine X! Tandem. At the time, it was the first open source protein identification software project to become available (a previous
academic effort had been attempted but was shutdown because of patent concerns). The release of the X! Tandem code – and the subsequent release a few months later
of the NCBI-sponsored open source search engine OMSSA – ushered in the modern era of proteomics informatics and computational biology.
Data set of the week: (2013/3/28)
Rapid phosphoproteomic and transcriptomic changes in the rhizobia-legume symbiosis. Overall rating: excellent data (worth study)
 This data set consisted of 382 results,
each representing a fraction from multidimensional chromatography experiments, using iTRAQ derivatives for quantitation.
The data files were made available through PASSEL (PASS00056).
It was published by
Rose CM, Venkateshwaran M, Volkening JD, Grimsrud PA, Maeda J, Bailey DJ, Park K, Howes-Podoll M, den Os D, Yeun LH, Westphall MS, Sussman MR, Ané JM, and Coon JJ in
Mol Cell Proteomics 2012 11:724-44 (PubMed).
This data set consisted of 382 results,
each representing a fraction from multidimensional chromatography experiments, using iTRAQ derivatives for quantitation.
The data files were made available through PASSEL (PASS00056).
It was published by
Rose CM, Venkateshwaran M, Volkening JD, Grimsrud PA, Maeda J, Bailey DJ, Park K, Howes-Podoll M, den Os D, Yeun LH, Westphall MS, Sussman MR, Ané JM, and Coon JJ in
Mol Cell Proteomics 2012 11:724-44 (PubMed).
This study represented the first major effort to characterize the proteome of the model plant species Medicago
truncatula, a legume species in the same genus as the commercially important forage crop Medicago sativa. The
study takes advantage of the known genome of the organism and provides some interesting insights into the role of protein
phosphorylation in metabolic processes associated with interaction between the plant and the symbiotic nitrogen-fixing bacteria that
it hosts in root nodules. The data was of excellent quality and it provides the best resource available for obtaining
phosphodomain annotation for legume proteins.
Data set of the week: (2013/3/21)
Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Overall rating: very good data (specialist interest)
 This data set consisted of 288 results,
each representing a single affinity purification experiment.
The data files were made available through PASSEL (PASS00117).
It was published by
Varjosalo M, Sacco R, Stukalov A, van Drogen A, Planyavsky M, Hauri S, Aebersold R, Bennett KL, Colinge J, Gstaiger M and Superti-Furga G in
Nat Methods 2013 Mar 3 (PubMed).
This data set consisted of 288 results,
each representing a single affinity purification experiment.
The data files were made available through PASSEL (PASS00117).
It was published by
Varjosalo M, Sacco R, Stukalov A, van Drogen A, Planyavsky M, Hauri S, Aebersold R, Bennett KL, Colinge J, Gstaiger M and Superti-Furga G in
Nat Methods 2013 Mar 3 (PubMed).
These experiments represent probably the best resource available for data that can be
used to teach the practice of computational biology as it applies to proteomics. The raw data, as well as the
information generated by two different peptide identification systems, can be directly downloaded and used.
The preliminary analyses available (on which the manuscript was based)
provide a nice insight into the current generic practices used for peptide and protein identification by many laboratories.
Given the data and information available in these files, it is practical to create a series of tutorial subsets of the files that
can be manipulated directly by students to answer many questions relevant to practical proteomics data analysis, for example:
Data set of the week: (2013/3/14)
Physiological Adaptation of the Rhodococcus jostii RHA1 Membrane Proteome to Steroids as Growth Substrates. Overall rating: excellent data (worth study)
 This data set consisted of 220 results
consisting of multidimensional chromatography single HPLC runs and experiment summaries.
The data files were made available through Proteomexchange (PXD000016).
It was published by
Haussmann U, Wolters DA, Fraenzel B, Eltis LD, and Poetsch A. in
J Proteome Res. 2013 12:1188-98 (PubMed).
This data set consisted of 220 results
consisting of multidimensional chromatography single HPLC runs and experiment summaries.
The data files were made available through Proteomexchange (PXD000016).
It was published by
Haussmann U, Wolters DA, Fraenzel B, Eltis LD, and Poetsch A. in
J Proteome Res. 2013 12:1188-98 (PubMed).
The organism being studied here (Rhodococcus jostii) belongs to a genus of
largely non-pathogenic prokaryotes belonging to the large suborder Corynebacterineae
that contains Mycobacteria and Corynebacteria. It is known to be very good at utilizing a wide variety of
substrates for growth, including many types of aromatic compounds that are not easily metabolized by
other bacteria. This study does an excellent job of characterizing changes in the membrane proteins present in
the organism in response to changing its carbon source from mainly pyruvate to cholesterol and cholate. The
experiments were consistently well done and the results provide an excellent resource for investigating
how well membrane proteins can be recovered using standard proteomics techniques. An interesting
feature of the results is the apparent presence of a threonine/serine specific, protein-N-acetyl transferase,
which results in protein N-terminal acetylation at threonine residues (with reduced activity for serine).
 The latest version of X! Tandem 2012.02.01 has completed its on-line testing and should be considered
a stable release. This new version of the open source search engine has several new features, including enhanced compatibility with
the SRM/MRM project Skyline (thanks to Brendan Maclean
and Brett Phinney). It also has a new sequence stacking mechanism that effectively produces non-redundant
sequence databases on the fly, which can significantly improve performance when using the proteomes of
multiple strains of prokaryote species or eukaryote protein sequences that correspond to multiple alternate
splice variants that are only different in the mRNA's UTRs.
The latest version of X! Tandem 2012.02.01 has completed its on-line testing and should be considered
a stable release. This new version of the open source search engine has several new features, including enhanced compatibility with
the SRM/MRM project Skyline (thanks to Brendan Maclean
and Brett Phinney). It also has a new sequence stacking mechanism that effectively produces non-redundant
sequence databases on the fly, which can significantly improve performance when using the proteomes of
multiple strains of prokaryote species or eukaryote protein sequences that correspond to multiple alternate
splice variants that are only different in the mRNA's UTRs. The main GPM system has been updated to use the latest version of the human proteome — ENSEMBL v. 70.37 —
which was based on the human genome sequence GRCh37.p10, Feb 2009. All of the relevant resources
(including annotated spectral library and proteotypic peptides) have been updated to the new sequence set.
The annotation file for human SNAPs (Single Nucleotide Amino acid Polymorphisms) has been updated to dbSNP 137 (1,690,969 SNAPs),
using ENSEMBL's Biomart interface for the DNA-to-protein coordinate and allele mapping.
The main GPM system has been updated to use the latest version of the human proteome — ENSEMBL v. 70.37 —
which was based on the human genome sequence GRCh37.p10, Feb 2009. All of the relevant resources
(including annotated spectral library and proteotypic peptides) have been updated to the new sequence set.
The annotation file for human SNAPs (Single Nucleotide Amino acid Polymorphisms) has been updated to dbSNP 137 (1,690,969 SNAPs),
using ENSEMBL's Biomart interface for the DNA-to-protein coordinate and allele mapping.
 The Journal of Proteome Research has released it's special issue
associated with the launch of the chromosome-based Human Proteome Project. This issue is freely
available on-line and it includes articles from many of the cHPP national groups, as well as
technical papers associated with the creation of boutique databases, laboratory techniques
and visualization systems specifically designed for use by the cHPP. There are also several general
articles on the current status of experimental evidence for the existence of human proteins, discussing
the gaps in our knowledge at the moment.
The Journal of Proteome Research has released it's special issue
associated with the launch of the chromosome-based Human Proteome Project. This issue is freely
available on-line and it includes articles from many of the cHPP national groups, as well as
technical papers associated with the creation of boutique databases, laboratory techniques
and visualization systems specifically designed for use by the cHPP. There are also several general
articles on the current status of experimental evidence for the existence of human proteins, discussing
the gaps in our knowledge at the moment.
Copyright © 2013, The Global Proteome Machine Organization
|