Data sets of the year: (2010/12/26) Technical, Biological and Clinical.
This week we are awarding the title "Data set of the year" to three outstanding
examples of publicly available proteomics experimental data. These awards are in three
categories:
The ENSEMBL proteome for M. musculus (mouse) has been updated to the latest version
(ENSEMBL v.60, NCBIm37).
The proteome is broken down into an individual file for each chromosome (1-19, X, Y, MT and other),
each of which is also available by FTP. This division
of protein sequences makes it possible to create individual proteomes for male and female, which can be searched individually
on any of the GPM public servers.
The individual Annotated Spectrum Libraries for each human and mouse chromosome have been made
available in the human_chromosomes and
mouse_chromosomes directories
of the GPM FTP site. The P3 algorithm peptide contigs are also available via FTP.
Data set of the week: (2010/12/19) An Expanded Oct4 Interaction Network: Implications for Stem Cell Biology, Development, and Disease.
This study contains 7
LC/MS/MS runs, from pull-down experiments.
The manuscript describing this work was published by
Pardo M, Lang B, Yu L, Prosser H, Bradley A, Babu MM, and Choudhary J,
Cell Stem Cell. 2010 6:382-95
(PubMed).
This study contains very high-quality pull-down results that represent
rarely observed Mus musculus proteins and peptides. Unfortunately, the original
data was not made publicly available: only spectra that resulted in identifications were
stored in PRIDE. Hopefully the authors will make the original data available at some
point so that a more thorough analysis can be performed.
Nota bene: In looking through these results, some may notice that there was no
observation of a protein named "Oct4". This seemly odd fact was due to
the confusing nature of protein naming: "Oct4" is not a currently accepted name
for any mouse protein. The current name for that gene product is "Pou5f1" (POU domain, class 5, transcription factor 1),
corresponding to ENSMUSP00000025271.
Inspection of the current observations show clearly that this protein has been
over-represented in samples coming from mouse embryonic stem cells.
Update of the H. sapiens proteome (2010/12/16)
The ENSEMBL proteome for H. sapiens (human) has been updated to the latest version
(ENSEMBL v.60, GrCH37.p2).
For the first time, the proteome is broken down into an individual file for each chromosome (1-22, X, Y, MT and other),
each of which is also available by FTP. This division
of protein sequences makes it possible to create individual proteomes for male and female, which can be searched individually
on any of the GPM public servers.
Increased search capacity for "human" (2010/12/14)
Because of an increase in utilization of the "human" search server (human.thegpm.org),
we have tripled the number of CPU's available for searches using the FASTA sequence lists
available from that server.
Data set of the week: (2010/12/12) Nucleosome-interacting proteins regulated by DNA and histone methylation.
This study contains 160
LC/MS/MS runs, grouped into sets of SDS-PAGE bands.
The manuscript describing this work was published by
Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, and Kouzarides T,
Cell 2010 143:470-84
(PubMed).
This work demonstrates the extent to which SILAC quantitation has become a main stream
technique in molecular biology. The study addresses a biologically important question,
uses an exellent lab to perform the proteomics instrumental analysis and applies
straightforward, established informatics methods to interpret the proteomics data in the context of
the biological question.
Data set of the week: (2010/12/05) Comparative shotgun proteomics using spectral count data and quasi-likelihood modeling.
This study contains 153
LC/MS/MS runs, grouped into sets of MudPit experiments. The analysis for each individual LC/MS/MS and
summaries of the MudPit runs were recorded.
The manuscript describing this work was published by
Li M, Gray W, Zhang H, Chung CH, Billheimer D, Yarbrough WG, Liebler DC, Shyr Y, and Slebos RJ,
J Proteome Res. 2010 9:4295-305
(PubMed).
While this set of data was generated for a specific statistical study, it also represents a very good
resource for anyone interested in the study of signal analysis, bioinformatics or statistics as they relate to proteomics
experimental analysis. The tissues selected were of clinical interest (head and neck carcinomas), the
equipment was state-of-the-art and the experimental groups involved were first rate. Many data sets generated
for bioinformatics analysis are not really representative of current best laboratory practices, but this one genuinely
exceeds expectations.
Full database dumps available on TRANCHE (2010/12/03)
Periodically, we make the complete set of databases that make up GPMDB available on
TRANCHE. The version now available was this week's backup, dated 2010/11/28, which
you can download from this link.
The FTP version of these files is
also available from the GPM FTP site here.
The files were generated using the mysqldump utility
and they can be used to generate the three databases used to create GPMDB displays:
GPMDB - the main record of the proteomics data;
ENSPMAPDB - protein accession numbers and descriptions; and
PEAKDB - the database used to generate MRM and spectrum library information.
New GPMDB Wiki server (2010/11/30)
We are in the process of moving the GPMDB Wiki from its old server to a new one. The new
server should be considerably faster than the old one and it has some features that make
it easier for us to work with. It may take a day or two for the Internet DNS system to
catch up with this change, so if you are adding material to the Wiki, please wait until
Thursday to make your changes.
Data set of the week: (2010/11/28) Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics.
This study contains 28
tissue sample data sets.
The manuscript describing this work was published by
Baerenfaller K, Grossmann J, Grobei MA, Hull R, Hirsch-Hoffmann M, Yalovsky S, Zimmermann P, Grossniklaus U, Gruissem W, and Baginsky S,
Science 2008 320:938-41
(PubMed).
This work is still probably the most comprehensive proteomics study of Arabidopsis thaliana tissues
available. Each of the individual samples corresponds to > 9,000 peptide identifications and > 1,000
non-redundant protein identifications. It can be used as a reliable catalogue of observable
peptides and proteins for the corresponding A. thaliana tissues and cell-culture samples.
Data set of the week: (2010/11/21) Prioritization of candidate protein biomarkers from an in vitro model system of breast tumor progression toward clinical verification.
This study contains 5
individual LC/MS/MS runs.
The manuscript describing this work was published by
Lau TY, Power KA, Dijon S, de Gardelle I, McDonnell S, Duffy MJ, Pennington SR, and Gallagher WM.,
J Proteome Res. 9(3):1450-9
(PubMed).
The data is a good example of what can be achieved using a QTOF-style instrument for analyzing
gel bands. The relatively good resolution obtained on the fragment ions makes peptide identifications more
positive (FDR ≈ 0.1%) and generally improves the confidence of the resulting protein identifications.
The approach used in the paper has some merit for determining the suitability of proteins
as biomarkers, although much of the comparitive work could have been done using
existing databases of observable plasma and serum proteins.
GPMDB Guide to the Mouse Proteome (2010/11/17)
In response to the positive reception that the GPMDB Guide to the Human Proteome
has received, we have constructed a similar set of spreadsheets for the Mus musculus proteome. You
can access the GPMDB Guide to the Mouse Proteome
as either an Excel spreadsheet or using a set of web pages that contain the same information, with
some additional hyperlinks to the original data used to construct the Guide.
Data set of the week: (2010/11/14) Proteomic Analysis of Human Nail Plate.
This study contains 40
individual LC/MS/MS runs.
The manuscript describing this work was published by
Rice RH, Xia Y, Alvarado RJ, and Phinney BS,
J Proteome Res. 2010 Nov 1
(Epub ahead of print, PubMed).
The data investigates the proteins present in two common but sparsely investigated
human tissues: hair and nail plate. These non-cellular tissues are composed mainly of
high-sulphur (hard) keratins and keratin-associated proteins in different proportions. These
proteins are unusually abundant on Chromosome 17, with more than 60 genes clustered between
chromosome coordinates 38,810,917-39,780,829 (see the Human
Proteome Guide for the gene names, positions and frequency of observation).
System problems because of power failure (2010/11/07)
A power failure at one of our main sites may lead to problems accessing some features of GPM until
tomorrow morning (Pacific Time), Nov. 8.
Data set of the week: (2010/11/07) Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate.
This study contains 24
individual result sets derived from SDS-PAGE gel bands.
The manuscript describing this work was published by
Lowery DM, Clauser KR, Hjerrild M, Lim D, Alexander J, Kishi K, Ong SE, Gammeltoft S, Carr SA, and Yaffe MB
in EMBO J. 2007 26:2262-73
(PubMed).
This study demonstrates the power of protein affinity methods for enriching relatively rare, but biologically
important proteins. The result sets contain many of the best identifications observed for
proteins such as GRIPAP1, ROCK2, ANLN, EPB41L3, CLIP2 and the minichromosome maintenance complex. The methodology
used here was relatively simple, but it revealed an interesting, high quality interactome that will
take years of biological research to thoroughly investigate and understand.
Data set of the week: (2010/10/31) Genome analysis and genome-wide proteomics of Thermococcus gammatolerans, the most radioresistant organism known amongst the Archaea.
This study contains 7
individual result sets; each set is the union of all spectra collected from a single SDS-PAGE gel.
The manuscript describing this work was published by
Zivanovic Y, Armengaud J, Lagorce A, Leplat C, Guérin P, Dutertre M, Anthouard V, Forterre P, Wincker P, and Confalonieri F.
in Genome Biol. 2009;10(6):R70
(PubMed).
This study was a straightforward analysis of the proteome of a previously unexamined archaeon,
T. gammatolerans. What set this study apart was the level of competence displayed by the
research team in obtaining this data. The methodology used was straightforward, but they were
able to consistently generate spectra good enough so that ~50% of the spectra resulted in high quality identifications.
Generally, this type of strategy results in
high levels of human keratins 1, 2, 9 and 10 identified, but not in this case. The data corresponded to
>1000 T. gammatolerans proteins, with the largest of the individual gel sets having >60,000 identified peptides.
Data set of the week: (2010/10/24) Feasibility of large scale phosphoproteomics with HCD fragmentation.
This study contains 25
individual samples, contrasting two methods for phophopeptide detection.
The manuscript describing this work was published by
Nagaraj N, D'Souza RC, Cox J, Olsen JV, and Mann M
in J. Proteome Res. 2010 (Epub ahead of print,
PubMed).
This data set is a major game-changer for any group interested in high-throughput phosphopeptide detection.
The combination of HCD fragmentation with high accuracy parent and fragment ion
mass measurement described in the associated publication result a level of sequence
and PTM assignment accuracy that simply cannot be matched by the conventional
CID approach using a low accuracy LTQ for fragment ion analysis. It is
also clearly superior to ETD for high throughput phosphoproteomics: the physical
chemistry of ETD make it much better suited to the detailed characterization of difficult
cases rather than broad surveys of large mixtures.
Data set of the week: (2010/10/17) Coupled global and targeted proteomics of human embryonic stem cells during induced differentiation.
This data set contains 18
sample analyses.
The manuscript describing this work was published by
Yocum AK, Gratsch TE, Leff N, Strahler JR, Hunter CL, Walker AK, Michailidis G, Omenn GS, O'Shea KS, and Andrews PC
in Mol Cell Proteomics 2008 7:750-67
(PubMed).
This study utilizes MALDI TOF-TOF technology to provide an excellent survey of proteins
in embryonic stem cells. While MALDI has become a secondary ionization method compared
with electrospray, it still is a robust method for protein identification and it provides
the most reliable source for library spectra of singly charge peptide ions.
Data set of the week: (2010/10/10) Glycosylation signatures in Drosophila: fishing with lectins.
This data set contains 1
LC/MS/MS result.
The manuscript describing this work was published by
Vandenborre G, Van Damme EJ, Ghesquière B, Menschaert G, Hamshou M, Rao RN, Gevaert K, and Smagghe G.
in J Proteome Res. 2010 9:3235-42
(PubMed).
A carefully selected set of lectins was used to purify glycoproteins by affinity capture from
Drosophila melanogaster samples. The results show that this method was able to obtain
an unusually high quality set of identifications for proteins of this species, as demonstrated by the
very large fraction of "best ever" identifications for the proteins reported. The peptides
identified also show significantly more chymotryptic peptide cleavage than would be typical for such a study.
Maintenance outage at UK ENSEMBL (2010/10/05)
Because of a maintenance issue at the UK ENSEMBL site, GPM has switched
to using the USWest ENSEMBL site until the problem in England has been resolved.
Operationally, the USWest site is a complete mirror of the UK site, so no
changes in annotation or functionality should be caused by this switch-over.
Data set of the week: (2010/10/03) Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling.
This data set contains 1
LC/MS/MS result.
The manuscript describing this work was published by
Xu G, Paige JS, and Jaffrey SR
in Nat Biotechnol. 2010 28:868-73
(PubMed).
This data was obtained from a very interesting study that describes the utility of an
immunoaffinity method for purifying the peptides generated by the trypsin digest of
proteins that have N-lysyl-ubiquitination. Trypsin cleaves away most of the ubiquitin bound
to the lysine sidechain, leaving a Gly-Gly sequence attached. By generating an antibody that
was specific for this type of modified lysine sidechain, they were able to isolate peptides
from ubiquitinated proteins. This purification allowed them to overcome the large concentration ratio between the modified
and unmodified proteins that has made identifying this type of modification difficult in the past.
The availability of this antibody should make many interesting
studies of the ubiquitin-mediated protein degradation pathway possible.
Data set of the week: (2010/09/26) The Asia Oceania Human Proteome Organisation Membrane Proteomics Initiative. Preparation and characterisation of the carbonate-washed membrane standard.
This data set contains 2
LC/MS/MS results.
The manuscript describing this work was published by
Peng L, Kapp EA, Fenyö D, Kwon MS, Jiang P, Wu S, Jiang Y, Aguilar MI, Ahmed N, Baker MS, Cai Z, Chen YJ, Van Chi P, Chung MC, He F, Len AC, Liao PC, Nakamura K, Ngai SM, Paik YK, Pan TL, Poon TC, Hosseini Salekdeh G,
Simpson RJ, Sirdeshmukh R, Srisomsap C, Svasti J, Tyan YC, Dreyer FS, McLauchlan D, Rawson P,
and Jordan TW.
in Proteomics. 2010 May 18
(PubMed).
This study, the results of a HUPO-affiliated AOHUPO project, demonstrate the effectiveness of
a standardized, relatively simple protocol for the enrichment of membrane proteins. A quick
inspection of the GO displays for
unwashed and
carbonate washed samples
proves this point very nicely. Many groups still seem to believe that membrane proteins are
difficult to observe using proteomics methods, so a straightforward study such as this one
demonstrating the contrary is a welcome addition to the field and an excellent subject for
a HUPO study.
Data set of the week: (2010/09/19) Plasticity and robustness of protein patterns during reversible development in the honey bee (Apis mellifera).
This data set contains 23
LC/MS/MS results. The original data was obtained from Peptidome (Study PSE129).
The manuscript describing this work was published by
Wolschin F, and Amdam GV
in Anal Bioanal Chem. 2007 389:1095-100
(PubMed).
This study explores the protein levels associated with behavioral differences in
honeybees. Apis mellifera is an economically important species with a complete genome
but which has recieved only limited attention from the proteomics community. Fortunately
bee proteomics scientists have been very active in contributing their data to public
repositories. Inspection of the
list of all A. mellifera proteins
in GPMDB shows that more than 2450 proteins have been observed and a surprising number of them have been
observed more than 500 times.
Data set of the week: (2010/09/12) Identification of pathways associated with invasive behavior by ovarian cancer cells using multidimensional protein identification technology (MudPIT).
This data set contains 252
LC/MS/MS results. The original data was obtained from TRANCHE.
The manuscript describing this work was published by
Sodek KL, Evangelou AI, Ignatchenko A, Agochiya M, Brown TJ, Ringuette MJ, Jurisica I, and Kislinger T.
in Mol Biosyst. 2008 4:762-73
(PubMed).
This study contains probably the best information set for the detailed exploration of proteomics as
a reproducible technology. Six different ovarian cancer cell lines were examined, each of which
is analyzed in six replicates, each replicate containing six SCX fractions. While this study was designed
to explore the differences between these cell lines, it also affords a truly useful collection of
data for anyone interested in proteomics
sample preparation reproducibility, measurement undersampling, search engine effectiveness, peak finding
efficacy or any other aspect of proteomics data generation and handling.
The GPM results are grouped according to cell line replicates, with each replicate having six entries
corresponding to the individual SCX fractions, followed by a summary result generated from those
six analyses. A description containing a statement like "Data directory: SKOV_5" indicates that
the result was obtained from replicate "5" of cell line "SKOV".
1,500,000,000 spectra analyzed (2010/09/07)
Some time yesterday (Sept. 6, 2010) the Global Proteome Machine processed its
1,500,000,000th spectrum. We would like to thank all of the direct contributors
to this project, as well as all of the investigators who have made there data available
through TRANCHE, PRIDE and Peptidome.
Data set of the week: (2010/09/05) A quantitative proteomics design for systematic identification of protease cleavage events.
This data set contains three (3)
COFRADIC analyses (COmbined FRActional DIagonal Chromatography).
The original manuscript describing this work was published by
Impens F, Colaert N, Helsens K, Ghesquiere B, Timmerman E, De Bock PJ, Chain BM, Vandekerckhove J, and Gevaert K
in Mol Cell Proteomics. 2010 Jul 13
(PubMed).
The study demonstrates a relatively straightforward method for determining the
cleavage specificity of proteolytic enzymes. The data analysis technique used in the
original paper is somewhat complex, but the more flexible modes of analysis available
in the GPM simplied the process considerably. Simple inspection of the AAA display allows
the assignment of the appropriate cleavage specificities for the enzymes:
A novel parser to display X! series data output has been developed by Thilo Muth, Marc Vaudel,
Harald Barsnes, Lennart Martens, and Albert Sickmann and published in Proteomics 2010, 10:1522-4
(PubMed). This
parser allows the user to browse through results files and generate annotated diagrams
of the individual spectra that support particular peptide sequence assignments. It is
a nice, cross-platform implementation and doesn't require any additional software (other than
Java) to run.
mzML 1.1.0 compatibility (2010/08/31)
Thanks to a code contribution from Fredrik Levander from the
Department of Immunotechnology - Protein Technology at Lund University,
X! Tandem, P3, Hunter and Common have been updated to deal with the latest version of mzML. The
code repository has the appropriately altered version of the source code files
saxhandler.cpp;
saxmzmlhandler.cpp; and
saxmzmlhandler.h.
The code in the release versions will be changed in the next full release of the software. All
of the public GPM search sites have been updated and people with GPM-XE can obtain the mzML
compatible executables using the normal software updating link.
Request For Comment 2010.08.30: changes to cRAP
The common Repository of Adventitious Proteins project (cRAP) would like to
request user comments on a proposed revision of the protein sequences currently
included in its list of proteins. If you would like to comment, please email
your suggestions to rfc@thegpm.org.
This RFC will remain active until October 1, 2010, at which time the resulting
changes will be posted on the new RFC page.
The proposed changes are as follows:
The removal of the "category 4" proteins from the list
(see the cRAP page for a complete list of these
proteins). The affected sequences (the Sigma/Aldrich Universal Protein Standard) were
originally place in cRAP in anticipation of broad use of this mixture resulting in
significant levels of contamination of non-human samples. Our analysis of the
data shows that this has not happened. The continued presence of these
alternate protein sequences in cRAP has given rise to added complexity in human
data samples, which we feel it would be best to eliminate.
The addition of two new viral protein sequences. These sequences, the heavy and light
chain of the human adenovirus C E1B control protein, are observed in proteomics
data obtained from HEK293 cells. These proteins are not the result of an infection: they
are caused by the DNA insertion that immortalized this cell line. These proteins have no
significant homology to any non-viral proteins and their inclusion should not increase
the complexity of analyzed results.
The addition of one new bacterial protein, the DNA K chaperonin from common cell culture
infectious agent Mycoplasma hominis. This bacteria can be present at some level
in cell culture and this protein is meant serve as an indicator of the potential
infection of any sample derived from these cells. M. hominis ATP synthase subunit beta is another potential
candidate, however its sequence has sufficient homology to mammalian ATP synthase that
the danger of false positive assignments makes it a less attractive candidate marker.
Data set of the week: (2010/08/29) Human Ccr4-Not complexes contain variable deadenylase subunits.
This data set contains nine (9)
LC/MS/MS analyses.
The original manuscript describing this work was published by
Lau NC, Kolkman A, van Schaik FM, Mulder KW, Pijnappel WW, Heck AJ, and Timmers HT.
in Biochem J. 2009 422:443-53
(PubMed).
The study contained eight (8) pulldown experiments and one (1) control. Each
pull-down is annotated with the bait protein. The experiment uses the combination of Lys-C and
bovine trypsin characteristic of the Heck group, which generates a rather
complete set of trypsic peptides, although there were a signficant number of
non-tryptic peptides generated. The sample preparation method used urea, so there
was also a significant number of carbamylated peptides detected. Neither of these artifacts
affect the conclusions of the study.
The study contains also contains a surprising number of
protein identifications that are the best so far obtained in GPMDB, e.g., TNKS1BP1, RAVER1,
FHL2, RQCD1, RNF219, UBAP2L, BAG3 as well as the bait CNOT proteins. Pull-down experiments,
with their ability to purify an unusual fraction of proteins, seem to be very effective
at obtaining the best observations of rare proteins, compared to large MudPit-style survey
experiments.
Data set of the week: (2010/08/22) Low abundance proteome of human red blood cells captured by combinatorial peptide libraries. Behavior of mono- to hexapeptides.
This data set contains 19
LC/MS/MS analyses.
The original manuscript describing this work was published by
Sim C, Bachi A, Cattaneo A, Guerrier L, Fortis F, Boschetti E, Podtelejnikov A, and Righetti PG.
in Anal Chem 2008 80:3547-56
(PubMed).
This study is an excellent example of a very important class of study: attempting to
use novel separation strategies to increase the dynamic range of tissue proteomics. The
particular strategy used in this case appears to work quite well at obtaining distributions
of proteins with limited specificity, while at the same time producing fractions depleted in high abundance
proteins. Technically, the data is also very quality and it contains an unusual number of
high confidence identifications of relatively small peptides (< 1000 Da).
ASLs for A. mellifera, A. carolinensis, X. tropicalis and X. laevis available (2010/08/18)
We are happy to be able to make available the first Annotated Spectrum Libraries for the
honeybee (Apis mellifera),
a lizard (Anolis carolinensis) and two amphibians (Xenopus tropicalis and Xenopus laevis).
These libraries are now available for download (FTP)
or for on-line searches at the X! Hunter search server.
Data set of the week: (2010/08/15) Quantitative analysis of kinase-proximal signaling in lipopolysaccharide-induced innate immune response.
This data set contains 73
LC/MS/MS data sets of obtained from mouse RAW 264.7 cells (macrophage cell line) that have been treated with lipopolysaccharide
to simulate infection with Gram-negative bacteria.
This data was published by
Sharma K, Kumar C, Kéri G, Breitkopf SB, Oppermann FS, and Daub H in J Proteome Res. 2010, 9:2539-49
(PubMed).
The goal of the paper was to follow TOLL-like receptor phospho-signaling during this sort of
simulated infection using SILAC: a combination of unlabelled and labelled samples with
two different isotopic tag pairs (K(4),R(6) and K(8)R(10)) were used to detect differential
protein and phosphopeptide concentrations.
In addition to the biological conclusions, this data contains some excellent examples of a common
analytical artifact associated with the use of titanium dioxide phosphopeptide enrichment. Metal oxide
columns work by binding peptides with low pIs (i.e., acidic peptides). While phosphopeptides certainly
fill the bill as being acidic relative to most peptides, normal peptide sequences with multiple acidic sidechains are
also strongly enriched by these columns. This effect can be clearly seen by using the pI vs. RT
and the amino acid analysis
displays. In example used here, most of the peptides detected have a pI < 5. Aspartic acid (D) and glutamic acid (E) residues in the detected peptides
are enriched to 250% and 215% of their expected composition, based on the composition of the associated proteins.
Release of the GPMDB Guide to the Human Proteome (2010/08/12)
The Human Proteome Organization (HUPO) is in the process of developing a Human
Proteome Project (HPP) that will be officially announced at the upcoming HUPO meeting in Sydney
this September. As our contribution to the planning process for this important initiative,
the information in GPMDB has been summarized into a collection of spreadsheets that we are calling the
GPMDB Guide to the Human Proteome. This
guide has the information organized into separate spreadsheets for each chromosome, as well as three transposons and
mitochrondrial DNA. The protein accession numbers, HGNC names and chromosomal coordinates were taken from ENSEMBL v. 58.
This first version of the Guide is available in the following formats:
Release 5 of NCTA tissue proteomics information available (2010/08/10)
The fifth release of the Normal Clinical Tissue Alliance
list of proteins found in normal clinical tissue samples is now available. The information can be accessed
through the NCTA HTML interface,
which also allows the download of speadsheets for all available tissues. This release is the first to include
lung (BTO:0000763), made possible by new data
from Dan Lieber's group at Vanderbilt University. The BTO
interface in GPMDB has also been updated with this new information.
Probing Proteins: the Scientist (2010/08/09)
This month's issue of "The Scientist" has a good general article
on the use of online proteomics data called Probing Proteins, written by Jeffrey M. Perkel. It is written for the
general biomedical science community with some explanations of how the different data
and information sources can be used.
Data set of the week: (2010/08/08) Comparative proteome profiling of Mycobacterium tuberculosis: the response of drug-resistant and drug-sensitive stains.
This data set contains 6 (six)
MudPit data sets of two different strains of M. tuberculosis, A12998 (daughter strain, drug-resistant) and A7494 (parent strain, drug-sensitive).
This data was published via upload to Peptidome
as Study PSE133 by
Moo-Jin Suh, Rembert Pieper, and Shih-Ting Huang from the J. Craig Venter Institute.
From Peptidome: The study describes the analysis of proteins from Drug-resistant and
-sensitive strains of Mycboacterium tuberculosis. LC-MS-based proteomics approach was combined with APEX to quantitatively measure relative proteins abundance and to compare the cellular protein composition of
Mycobacterium tuberculosis strains A12998 (daughter strain, drug-resistant) and A7494 (parent strain, drug-sensitive).
The results are probably the most thorough analysis of proteins from this important pathogen and
they make up a large fraction of the Annotated Spectrum Libraries available from M. tuberculosis
strains.
An unexpected piece of information made available through this data set is a good initial
measurement of the phosphoproteome of this prokaryote. M. tuberculosis is known to have a serine/threonine
kinase and this data set has a number of very good phophopeptides generated by this kinase. The kinase
appears to prefer threonine phosphorylation, with a S:T ratio of about 1:3. This ratio is the reverse
of typical eukaryote kinases, which seem to prefer serine by about 3:1. The phosphoproteome
generated from this study is available in either
Excel,
html or
tab-separated text formats, as projected on to the proteome of
strain CDC1551. Note: the original analysis in Peptidome did not include phosphorylation, so these
results are only present in the GPMDB re-analysis. It would be very useful to
have an IMAC-type study done on these and other M. tuberculosis strains.
Data set of the week: (2010/08/01) In-depth proteomic analyses of direct expressed prostatic secretions.
This data set contains 9 (nine)
MudPit data sets, each measured from a different prostatic fluid sample from individuals with prostate cancer.
The original raw data was obtained from TRANCHE.
It was published by Drake RR, Elschenbroich S, Lopez-Perez O, Kim Y, Ignatchenko V, Ignatchenko A, Nyalwidhe JO, Basu G, Wilkins CE, Gjurich B, Lance RS, Semmes OJ, Medin JA, and Kislinger T. in
J Proteome Res. 2010, 9:2109-16 (PubMed).
The results show the amount of variability that can be expected when analyzing biological replicates
of clinically sampled material. The identifications were very high quality and are the best quality measurements of
many rather rare proteins, such as KLK3 (prostate-specific antigen) and ACPP (Prostatic acid phosphatase). The data
shows moderate levels of carbamylation from the urea solublization method used. There were also significant concentrations of
peptides generated by non-tryptic cleavage, probably from the presence of proteases in the sample itself as the cleavage sites were
not chymotryptic. An examination of the AAA page (e.g., sample #2)
showed that the "Pre" and "C-terminal" columns were broadly populated for most residues,
not just the K and R residues normally expected in a trypsin cleavage experiment.
Interestingly for a sample obtained from prostate secretions, no proteins originating from genes on the Y chromosome
were detected. This fact points out a general feature of proteomics: there does not seem to be any "common sense"
association between tissue-specific protein concentrations and chromosomes.
Data set of the week: (2010/07/25) Proteomic analysis of the secretome of human umbilical vein endothelial cells using a combination of free-flow electrophoresis and nanoflow LC-MS/MS.
This data set contains a single
LC/MS/MS data set, using a combination of free-flow electrophoresis and nanoflow HPLC separations.
The original raw data was made available as a Scaffold file from a web site maintained by the authors (www.vascular-proteomics.com).
It was published by Tunica DG, Yin X, Sidibe A, Stegemann C, Nissum M, Zeng L, Brunet M, and Mayr M in
Proteomics. 2009, 9:4991-6 (PubMed).
This study attempts to discover a difficult thing: the secretome of human umbilical vein endothelial cells in the face
of the background proteins in a complex growth medium. The
results provide a good basis for the examination of this important cell type, with a very good set of
identifications that provides a broad survey of the proteins that can be readily obtained
from these cells.
Additions to the S. cerevisiae proteome (2010/07/21).
As has been mentioned several times in the Data Set of Week announcements, proteins from the two viruses
Saccharomyces cerevisiae virus L-A (L1) and Saccharomyces cerevisiae virus L-BC (La) have
been very commonly observed in proteomics data sets obtained from S. cerevisiae. The
signals associated with peptides from these viruses can be quite strong, so it is our belief that
not including the proteome of these viruses when searching S. cerevisiae data may lead to missed (or misinterpreted)
identifications. Therefore, the proteome of S. cerevisiae has been altered on all of the GPM public
search sites to include the proteomes of these viruses
(NC_003745 and
NC_001641) by default.
These two viruses contain a total of five (5)
proteins, so there will be little impact on the overall search speed caused by this proteome-level change.
New X! Hunter Annotated Spectrum Libraries for model species available (2010/07/20).
The most recent versions the the Annotated Spectrum Libraries for H. sapiens, M. musculus,
R. norvegicus, S. cerevisiea, D. melanogaster and C. elegans are now available
at the X! Hunter project ftp site. These
libraries use the ENSEMBL v. 58 protein sequences. Specifics of the build are available at the ASL
statistics page. The libraries are available for searches at the X! Hunter
server.
Data set of the week: (2010/07/18) Proteomics Analysis of the Causative Agent of Typhoid Fever.
This data set contains 313
LC/MS/MS runs using Thermo LTQ mass spectrometers.
The original raw files originally from the Resource Center for Biodefense Proteomics Research, which
has been superceded by the Pathogen Portal (raw data).
It was published by Ansong C, Yoon H, Norbeck AD, Gustin JK, McDermott JE, Mottaz HM, Rue J, Adkins JN, Heffron F, and Smith RD in
J Proteome Res. 2008, 7:546-57 (PubMed).
This very thorough data set is the primary large collection of information that has allowed for the
creation of the rather comprehensive annotated spectrum libraries that are now available for
S. enterica related species, including S. typhi and S. typhimurium. The Pacific Northwestern
National Laboratory group was an early proponent of making publicly-funded proteomics raw data widely available and
their efforts legitimized the idea for many other groups.
Data set of the week: (2010/07/11) Discovery of Anthrax Biomarkers Using Label-Free Quantitative Phosphoproteomics via Mass Spectrometry.
This data set contains 66 individual phosphopeptide enriched
LC/MS/MS runs made using a Thermo Orbitrap hybrid mass spectrometer.
The original raw files were transferred from TRANCHE.
The data was credited to Nathan P. Manes, Li Dong, Weidong Zhou, Xiuxia Du, Nikitha Reghu, Arjan C. Kool,
Dahan Choi, Charles L. Bailey, Emanuel F. Petricoin III, Lance A. Liotta, and Serguei G. Popov.
It was made available prior to publications, although some part of the data was presented at the 2010 ASMS conference.
The analyzed results are simply the best, most consistent set of phosphopeptide results that we have ever seen.
The combination of sample preparation, HPLC and mass spectrometry used by the authors has generated
what can only be considered a milestone in the application of phospho-proteomics technique to
real tissue samples.
X! Hunter Annotated Spectrum Libraries for 115 prokaryote and 4 virus proteomes are now available
The X! Hunter ASL collection of bacterial proteome libraries has been expanded from the original 16 species to now include 115 prokaryote species and strains.
The new species include important pathogenic organisms, such as Shigella dysenteriae, Yersinia pestis and Brucella abortus. The new release
also includes 30 individual strains of Escherichia coli. This update of the ASL collection has, for the first time, libraries for four viruses:
Data set of the week: (2010/07/04) Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions.
This data set contains 61 individual experiments using
both SILAC and label-free quantitation. The experimental protocols used either trypsin or endo-LysC to digest
the proteins, depending on the type of protocol being used.
The original raw files were transferred from TRANCHE.
The data was published by Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M in
J Cell Biol. 2010 189:739-54 (PubMed).
The data was generated to demonstrate the utility of a new technique for protein quantitation
developed by the authors: "quantitative BAC-green fluorescent protein interactomics" (QUBIC). The
technique is meant to be applied to the quantitative study of protein-protein interactions, several of
which are demonstrated here. The technical quality of the MS/MS data is excellent, with many ids for individual proteins
in the top 10% of all GPMDB observations.
Video tutorial (2010/07/03): Finding phosphorylation sites using GPMDB
We have started to make a set of tutorial videos to explain how to use GPMDB for common
biomedical research tasks. The first video in this set describes the steps necessary to
find the observed phosphorylation sites for a particular protein. The description is in
the form of a casual conversation between a biomedical researcher (as played by HRM Queen Elizabeth II) and
a GPMDB power user (Beavo the clown), during a chance meeting at the dépanneur. We will be releasing these videos as they
are produced. Please see our new tutorials page to
check for new videos.
Data set of the week: (2010/06/27) mTAL Phosphoproteome Data.
This data set contains metal oxide enriched LC/MS/MS observations
of phosphopeptides from R. rattus medullary Thick Ascending Limb (mTAL) cells.
The raw files were transferred from TRANCHE.
The original analysis was reported by Ruwan Gunaratne, Guozhong Ma, Trairak Pisitkun, and Mark A. Knepper as part of the mTAL-PD
project. It appears to be closely related to the Collecting Duct
Phosphoproteome Database.
The phosphorylated domains obtained are interesting because there is surprisingly little publicly available data from
rat cell lines or tissue samples. The phosphopeptide enrichment here was somewhat less effective
than in some other studies, however overall it is quite typical of IMAC phosphopeptide enrichment studies. This
study has significantly added to the known phosphorylated domains for available R. rattus through GPMDB's pSYT interface.
Data set of the week: (2010/06/20) Proteomic analysis of mouse brain microsomes: identification and bioinformatic characterization of endoplasmic reticulum proteins in the mammalian central nervous system.
This data set contains 1 2DLC MS/MS and 3 1DLC MS/MS rusn obtained
from mouse brain microsomal preparations. The original data was transferred from TRANCHE.
The original data analysis was reported by Stevens SM Jr, Duncan RS, Koulen P, Prokai L. in J Proteome Res. 2008 7:1046-54.
(PubMed).
This data set is interesting in a number of ways. It shows the difference in the depth of analysis available using
of multi-dimensional chromatographic analysis versus simple, single separation HPLC. The three repetitions of the
1D LCMS approach give a good indication of the statistical variability that is to be expected caused by the
under-sampling inherent in this type of measurement. A Gene Ontology analysis of the data (e.g., GPM33080005862)
shows the complexity of real microsomal samples, compared to simply believing that they contain only membrane and membrane-associated proteins.
A similar study can be compared, showing
some significant differences in microsome proteome composition, which are most likely due to variations in the
sample preparation methods.
Data set of the week: (2010/06/13) The minor salivary gland proteome in Sjögren's syndrome.
This data set contains 2 LC-MS-MS runs obtained
from human salivary gland tissue. The original data was transferred from PRIDE entries 7962-3.
The data was reported by Hjelmervik TO, Jonsson R, Bolstad AI. in Oral Dis. 2009 15:342-53.
(PubMed).
The two sets of identifications are meant to show the differences in the protein compliment of
salivary glands caused by the autoimmune disease, Sjögren's syndrome.
Technically, the data is a good example of the use of a high resolution MS/MS device (ESI-QTOF, Ultima Global) applied to
tissue samples. The high accuracy fragment ion masses significantly improve the quality of the
identifications.
Data set of the week: (2010/06/06) Identification of Ricin and Concanavalin A-binding Trypanosoma brucei Glycoproteins.
This data set contains 1 data set obtained
from T. brucei. The original data was transferred from PRIDE 9223.
A portion of the data was report by Izquierdo L, Schulz BL, Rodrigues JA, Güther ML, Procter JB,
Barton GJ, Aebi M, Ferguson MA in EMBO J. 2009 28:2650-61
(PubMed).
The data was obtained by using the the lectins concanavalin A and ricin to pull down glycoproteins from
T. brucei (blood stream form) and then glycosidases were used to remove the N-linked glycosylation, leaving
a deamidated asparagine residue behind. Any deamidated N residue that was associated with the N-{P}-[ST] glycosylation
motif should be considered a potential N-linked glycosylation site. You can see just these peptides by
clicking here.
Mouse protein phosphorylation sites (2010/06/05)
As a companion to the list of known human phosphorylation sites, we have also compiled a similar
list for the mouse proteome, based on the data in GPMDB.
This list is available in Excel spreadsheet,
tab-separated text
and HTML
formats. It contains 10,266 phosphorylation sites on 4,209 protein sequences, with the following composition:
serine: 5,406;
threonine: 1,617; and
tyrosine: 3,243
Each ENSEMBL splice variant protein accession number has a listing of all observed sites
in a single row, that looks like the following:
ENSMUSP00000028190
Abl1
Y[253]4
Y[393]9
T[394]6
Y[469]6
The columns have the following interpretation:
The ENSEMBL accession number for the protein splice variant;
The MGI gene name associated with that accession number: there may be many splice variants
with the same gene name; and
The phosphorylated residue in the notation "X[nnn]C", where "X" is the residue type,
"nnn" is the sequence position of the residue and "C" is a relative confidence number for the assignment (higher is better).
We have to again thank all of the data contributors who have made these comprehensive
lists possible. When using this type of information, please use normal caution. Click here
for our recommendations for using lists of site assignments.
Human protein phosphorylation sites (2010/06/04)
We have come up with a list of known human phosphorylation sites, based on the data in GPMDB,
filtered through the same curation and quality control process that is used to create the Annotated Spectrum
Library collection. This list is available in
Excel spreadsheet,
tab-separated text
and HTML
formats. It contains 28,089 phosphorylation sites on 10,670 protein sequences, with the following composition:
serine: 16,806;
threonine: 4,361; and
tyrosine: 6,922
Each ENSEMBL splice variant protein accession number has a listing of all observed sites
in a single row, that looks like the following:
ENSP00000344789
ACACA
S[66]6
S[117]7
S[350]6
Y[1190]7
The columns have the following interpretation:
The ENSEMBL accession number for the protein splice variant;
The HGNC gene name associated with that accession number: there may be many splice variants
with the same gene name; and
The phosphorylated residue in the notation "X[nnn]C", where "X" is the residue type,
"nnn" is the sequence position of the residue and "C" is a relative confidence number for the assignment (higher is better).
We have to thank all of the data contributors who have made this type of comprehensive
list possible. When using this type of information, please use normal caution. Click here
for our recommendations for using lists of site assignments.
Data set of the week: (2010/05/30) Use of fluorescence-activated vesicle sorting for isolation of naked2-associated, basolaterally-targeted exocytic vesicles for proteomic analysis.
This data set contains 6 experiments obtained
from C. familiaris and it is probably the best single data set we have in GPMDB from the domestic dog proteome. This work was transferred from TRANCHE
and it was published by Cao Z, Li C, Higginbotham JN, Franklin JL, Tabb DL, Graves-Deal R, Hill S, Cheek K, Jerome WG, Lapierre LA, Goldenring JR, Ham AJ, Coffey RJ.
in Mol. Cell. Proteomics 2008, 7:1651-67 (PubMed).
The individual experiments show how well fairly straightforward proteomics techniques can perform
on vesicular membrane proteins. They also demonstrate of the type of comprehensive results that can be obtained using a proteome
sequence that is almost completely the result of genome annotation.
Homo sapiens microbiome associated proteome available for searches (2010/05/29)
In honour of the Human Microbiome Project publication in Science,
we have compiled all of proteomes translated for the Human Microbiome Project and assembled them into a searchable
FASTA file. You can add all of these proteomes to your searches using either the normal or
human search pages (it is the first selection in the "prokaryotes" box).
Email should be OK (& a rendering change) (2010/05/26)
Our Email changes are complete, so all email should be OK as of today.
In order to maintain compatibility with the latest version of the web browser Chrome,
we've had to disable the 3D rendering of the protein coverage displays. Once we've figured
out how to deal with the changes in Chrome (or a new release of Chrome fixes the problem),
we will reinstate the 3D rendering.
Email changes (2010/05/25)
We are changing our email system, so for the next few days emails sent to "thegpm.org" addresses might
not be received. We are sorry for any inconvenience.
Data set of the week: (2010/05/23) A Global Protein Kinase and Phosphatase Interaction Network in Yeast.
This data set contains 450 pull-down experiments obtained
from S. cerevisiae. This work was transferred from TRANCHE
and it was published by Ashton Breitkreutz, Hyungwon Choi, Jeffrey R. Sharom, Lorrie Boucher, Victor Neduva, Brett Larsen, Zhen-Yuan Lin, Bobby-Joe Breitkreutz, Chris Stark, Guomin Liu, Jessica Ahn, Danielle Dewar-Darch, Teresa Reguly, Xiaojing Tang, Ricardo Almeida, Zhaohui Steve Qin, Tony Pawson,
Anne-Claude Gingras, Alexey I. Nesvizhskii, Mike Tyers Science 2010 328:1043-6.
Each of the individual results is annotated with the identity of the bait used in the pull-down
experiment. L-A and L-BC virus proteins are present in some of the pull-downs. The group did a remarkably
job at detecting phosphopeptides for a study that did not do any specific enrichment for these
peptides.
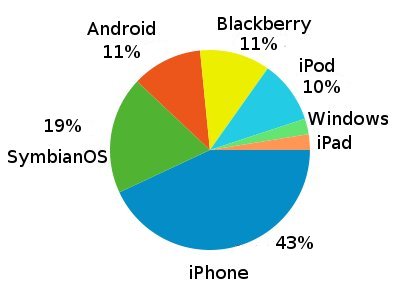
The iPhone wins (2010/05/20)
There are some people (probably with very good eye-sight) that use GPMDB on their mobile phones.
The chart below gives the breakdown of system usage by telephone operating system, showing that the three most
used mobile operating systems are
iPhone (Apple),
Symbian (mostly Nokia), and
a tie between Android (Google) and Blackberry (RIM).
Data set of the week: (2010/05/16) Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast.
This data set contains 505 LC/MS/MS runs obtained
from S. cerevisiae diploid and haploid populations. This work was transferred from TRANCHE
and it was published in de Godoy LM, Olsen JV, Cox J, Nielsen ML, Hubner NC, Fröhlich F, Walther TC, Mann M.
Nature. 2008 455:1251-4. (PubMed).
The results give a good indication of the relative abundance and observability of yeast proteins in
both haploid and diploid cells using either trypsin or endopeptidase LysC to generate peptides and SILAC labels to provide relative quantitation.
The data also shows very good examples of the major proteins observable
from the double stranded DNA viruses L-A and L-BC that are almost ubiquitously present in yeast cell cultures. In some
cases, these proteins are very strongly observed (e.g. protein #3 in GPM77711001229)
and the SILAC labelling can used to estimate the relative amounts of virus present in the two cell types. To located the virus and virus-related proteins in any
of the individual runs, type "virus" into the Find box at the top of any model page (click here for an
example).
Data set of the week: (2010/05/09) Phosphoproteome analysis of Drosophila melanogaster embryo.
This data set contains 24 LC/MS/MS runs obtained
from D. melanogaster embryos. This work was transferred from TRANCHE
and it was published in Zhai B, Villén J, Beausoleil SA, Mintseris J, Gygi SP,
J Proteome Res. 2008 7:1675-82 (PubMed).
The assignments in this data set give a good overview of phosphorylation in D. melanogaster and they
are good examples of phosphopeptides identified using an Orbitrap-LTQ hybrid instrument with CID. The mapped
phosphorylation sites from this data set were a major contribution to the pSYT annotation now available for
the fruit fly. The predominance of yolk proteins and other larvae-specific proteins in the identified peptides
gives a good view of the phosphorylation patterns on proteins that may be under-represented or absent from
studies that use mature flies or cells from tissue culture.
System change: discontinuation of IPI sequences (2010/05/03)
As mentioned in an earlier post, protein sequences using the
International Protein Index accession number scheme were discontinued in GPM search servers
as of May 1, 2010. The removal of this accession number system was made necessary because the
European Bioinformatics Institute (EBI), which originated IPI, has discontinued their support
for IPI sequences. All searches that have been performed using IPI accessions will still be
available and annotation for those searches will be maintained as long as possible. The ability to
convert ENSEMBL to IPI protein accession numbers will be maintained until ENSEMBL discontinues
its support for this type of conversion.
Data set of the week: (2010/05/02) Activated Macrophage Proteomics
This data set contains 9 merged results obtained
from human macrophages under various conditions. This work was transferred from a TRANCHE project
of the same name, created and maintained by Maureen M. Goodenow, Dept. of Pathology, Immunology and Laboratory Medicine
University of Florida.
The experiments reported by Dr. Goodenow are proteomics survey studies of macrophages, in which the
proteomes of treated cells are separated by SDS-PAGE and the resulting gel is sliced into 15 pieces. The
proteins are then digested, the peptides extracted and run using LC/MS/MS. Each one of the entries in GPMDB correspond
to the merged results of the 15 bands. They are good examples of what can be done using gel-slicing experiments to
obtain proteomics information about a cell type. It is also an admirable example of valuable data being made available to the
general community by an individual investigator.
Data set of the week: (2010/04/25) Large-scale quantitative LC-MS/MS analysis of detergent-resistant membrane proteins from rat renal collecting duct.
This data set contains 78 LC/MS/MS runs obtained
from membrance enriched fractions of tissue samples from rat renal ducts. It was originally published by
Yu MJ, Pisitkun T, Wang G, Aranda JF, Gonzales PA, Tchapyjnikov D, Shen RF, Alonso MA, Knepper MA.
in Am J Physiol Cell Physiol. 2008 295:C661-78
(PubMed).
The data was transferred to GPMDB from TRANCHE.
This study demonstrates that it is possible to generate very good results from membrane proteins isolated from tissue, even
those that do not readily dissolve in detergent solutions, such as lipid raft proteins. GO analysis of the
resulting protein identifications shows very significant enrichments in proteins known to be either integral
membrane, membrane associated or part of the extracellular matrix.
Data set of the week: (2010/04/18) Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins.
This data set contains 70 LC/MS/MS runs obtained
using TAP-tag protein isolation, SDS-PAGE separation followed by tandem mass spectrometry. It was originally published by Fernández E, Collins MO, Uren RT, Kopanitsa MV, Komiyama NH, Croning MD, Zografos L, Armstrong JD, Choudhary JS, Grant SG. Mol Syst Biol. 2009;5:269
(PubMed).
The data corresponds to the PeptideAtlas accession PAe001454 and was transferred to GPMDB.
The results are a good demonstration of the depth and detail of a particular molecular system that can be
obtained by coupling TAP-tagging with protein and subsequent peptide separations. The use of multiple gel
slices allows a depth of proteome coverage that would be difficult to obtain using other techniques.
Updated NCBI bacterial proteomes (2010/04/15)
The public GPM search servers have been updated with the most recent
set of bacterial proteomes available from the US National Center for Biotechnology Information.
This new set of sequences
adds approximately 200 new species to the list of proteomes available. Multiple
new species of human pathogens have been added, as well as additional strains of
species previously available.
ENSEMBL changes affecting GPM species (2010/04/13)
ENSEMBL has been widening its offerings of proteome sequences and annotation
over the last few months. Of the most utility to GPM has been the addition
of server systems to specifically deal with non-vertebrate species, such as
fungi, non-vertebrate metazoa, plants and protists. We are in the process of converting
some of the references and annotation sources in GPM to take advantage of
these new resources. To date, the following species have been switched to
using ENSEMBL metazoa:
A. gambiae,
C. elegans, and
D. melanogaster.
The following species have been switched to using ENSEMBL plant:
O. sativa,
A. thaliana, and
B. distachyon - this new proteome has been added to plant.thegpm.org.
The following species have been switched to using ENSEMBL fungi:
S. pombe.
Data set of the week: (2010/04/11) Proteomics of mouse liver microsomes
This data set contains 9 LC/MS/MS runs obtained
using SDS-PAGE separation followed by tandem mass spectrometry. It was originally published by Zgoda VG, Moshkovskii SA, Ponomarenko EA,
Andreewski TV, Kopylov AT, Tikhonova OV, Melnik SA, Lisitsa AV, and Archakov AI in
Proteomics, 2009,9:4102-5 (PubMed).
The data corresponds to the PRIDE accessions 8848-8856 and was transferred to GPMDB.
This data set is an example of the isolation of a specific experimental fraction (mouse liver microsome
from the endoplasmic reticulum) that provides a good representation of proteins not commonly observed, in
this case the cytochrome P450 family of metabolic oxidases. The quality of the isolation can be easily seen
when viewed as either KEGG pathways
or GO cellular components.
STRING-DB link for data models (2010/04/08)
STRING-DB is a long running project for the study of protein-protein interactions at a number of
different levels. A new link has been added to GPM "Main model display" pages to
make it easy for users to take advantage of the information in STRING-DB. The new link is
shown below:
As an example of the use of this new feature, try this
data set. Click on the "string-db" link of the display and then click the "Continue" button
on the data selection page generated by STRING-DB. You will then get a protein-protein interaction display
that indicates known interactions between the proteins that were discovered in the original data set. STRING-DB
has a number of interesting features for increasing or decreasing the stringency of the interactions displayed
as well as several different views on the data.
This feature is available on all data models that were constructed using ENSEMBL sequences.
Data set of the week: (2010/04/04) Use of shotgun proteomics for the identification, confirmation, and correction of C. elegans gene annotations
This data set contains 369 LC/MS/MS runs obtained
using a Thermo Finnigan LTQ instrument. It was originally published by Merrihew GE, Davis C, Ewing B, Williams G, Käll L, Frewen BE, Noble WS, Green P, Thomas JH, MacCoss MJ. in
Genome Res. 2008, 18:1660-9 (PubMed). The data
was obtained directly from the authors' web site
and it is not currently held in any of the other data sites.
The original analysis of this data set in the publication used the C. elegans WS150 proteome sequence and it was found to
indicate the presence of additional coding sequences. The analysis in GPMDB was performed using
the WS200 proteome (ENSEMBL v. 55), which has taken into account the original work. It serves
as a good example of the proteins that can be seen using conventional proteomics techniques in C. elegans.
IPI closure by European Bioinformatics Institute (2010/03/28)
The International Protein Index (IPI) will be closing later this year.
Because of this change, we will be discontinuing IPI protein sequences as an option effective May 1, 2010. All
existing IPI sequence searches will be retained in GPMDB and we will attempt to keep the
annotation information for these sequnces available as long as is practical. We would strongly
suggest that anyone who is currently using IPI should convert over to using ENSEMBL sequences as soon as
possible.
Data set of the week: (2010/03/28) Global proteomic profiling of Shigella dysenteriae Sd1617
This data corresponds to Peptidome Study PSE140, comprised of samples PSM1302,
PSM1303 and
PSM1304
The data was obtained by Rembert Pieper, Srilatha Kuntumalla, Shih-Ting Huang at the J. Craig Venter Institute
and it was transferred from Peptidome.
Each of the samples is composed of 3 replicate multidimensional chromatography runs of soluble
proteins obtained from S. dysenteriae. The tandem mass spectra are good quality, obtained using a Thermo LTQ
instrument. The results give a good indication of the type of depth and reproducibility that
can be expected in this type of straight-forward analysis of soluble proteins from an
enterobacterial culture.
Data set of the week: (2010/03/21) Global Impact of Oncogenic Src on a Phosphotyrosine Proteome
The data is composed of 31 separate runs.
The data was obtained from a study published in J. Proteome Res., 2008, 7 (8), pp 3447–3460, by Weifeng Luo, Robbert J. Slebos, Salisha Hill, Ming Li, Jan Brbek, Ramars Amanchy, Raghothama Chaerkady, Akhilesh Pandey, Amy-Joan L. Ham and Steven K. Hanks
(DOI: 10.1021/pr800187n). This information was
transferred from TRANCHE.
The data investigates the impact of Src transformation of mouse cells by determining the tyrosine phosphorylation
differences between control and transformed cells. The data also demonstrates the utility of using multiple peptidases
to increase the coverage of peptides, compared to trypsin alone. The data is very high quality LTQ data and it is an excellent
reference work for what is to be expected when looking for mouse tyrosine phosphophorylation.
Data set of the week: (2010/03/14) Quantitative phosphoproteomic analysis reveals vasopressin V2-receptor-dependent signaling pathways in renal collecting duct cells.
The data is composed of 2 separate sets, corresponding to the Peptidome accession numbers PSM1275 and
PSM1276. The data was
obtained from a study published in Proc Natl Acad Sci U S A. 2010 Feb 23;107(8):3882-7, by Rinschen MM, Yu MJ, Wang G, Boja ES, Hoffert JD, Pisitkun T, and Knepper MA
(PubMed). This information was
transferred from TRANCHE. The data is of high quality, containing good identifications of serine and threonine phosphorylation sites
in M. musculus proteins and it is an excellent example of the use of SILAC to monitor the relative quantitation of
protein phosphorylation.
Data set of the week: (2010/03/07) Phosphorylation dynamics during early differentiation of human emrbyonic stem cells.
The data is composed of 12 individual LC/MS/MS runs
obtained from a study published in Cell Stem Cell, Volume 5, Issue 2, 214-226, 7 August 2009 by Van Hoof D, Muñoz J, Braam SR, Pinkse MW, Linding R, Heck AJ, Mummery CL, and Krijgsveld J.
(PubMed). This information was
transferred from TRANCHE. Each of these data sets is large and contain significant
numbers of phosphorylated peptides.
The experiments performed were to investigate how "pluripotent stem cells self-renew indefinitely and possess characteristic
protein-protein networks that remodel during differentiation. How this occurs is poorly understood.
Using quantitative mass spectrometry, the (phospho)proteome of human embryonic stem cells (hESCs) was analyzed
during differentiation induced by bone morphogenetic protein (BMP) and removal of hESC growth factors."
Data set of the week: (2010/02/28) A Lectin HPLC Method to Enrich Selectively-glycosylated Peptides from Complex Biological Samples.
The data is composed of 83 individual LC/MS/MS runs
obtained from a study published in J Vis Exp. 2009 Oct 1;(32). pii: 1398 by Johansen E, Schilling B, Lerch M, Niles RK, Liu H, Li B, Allen S, Hall SC, Witkowska HE, Regnier FE, Gibson BW, Fisher SJ, and Drake PM
(PubMed). This information was
transferred from TRANCHE.
Briefly, plasma was depleted of the fourteen most abundant proteins using a multiple affinity removal system.
Depleted plasma was trypsin-digested and separated into flow-through and bound
fractions by SNA or AAL HPLC. The fractions were treated with PNGaseF to remove N-linked glycans,
and analyzed by LC-MS/MS on a QStar Elite. There is an accompanying video
explaining the methods used.
Data set of the week: (2010/02/21) Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors.
The data is composed of 729 individual LC/MS/MS runs
obtained from a study published in Nature Biotechnology by Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, and Drewes G.
(DOI: 10.1038/nbt1328). This information was
transferred from PRIDE (PRIDE accession numbers 2445-3178).
Labelling with iTRAQ is used for quantitative profiling of the consequences of the introductions of tge drugs imatinib (Gleevec),
dasatinib (Sprycel) and bosutinib in K562 cells confirms known targets including ABL and SRC family kinases.
Data set of the week: (2010/02/14) Cell-Specific Information Processing in Segregating Populations of Eph Receptor Ephrin-Expressing Cells.
This dataset was transfered to GPMDB via ProteoExchange from PRIDE.
The data is composed of 2 large LC/MS/MS runs
is from a study published in Science by Jørgensen C, Sherman A, Chen GI, Pasculescu A, Poliakov A, Hsiung M, Larsen B, Wilkinson DG, Linding R, and Pawson T
(DOI: 10.1126/science.1176615).
The data is from a set of quantitative mass spectrometric analyses of mixed populations of EphB2- and ephrin-B1–expressing cells that were labeled with different
isotopes revealed cell-specific tyrosine phosphorylation events. The data is of very high quality and it
has a very rich set of tyrosine phosphorylated peptides.
Data set of the week: (2010/02/07) The value of using multiple proteases for large-scale mass spectrometry-based proteomics.
This dataset was transfered to GPMDB via ProteoExchange from TRANCHE.
The data is composed of 15 LC/MS/MS runs
is from a study published in J. Proteome Research by Danielle L. Swaney, Craig D. Wenger and Joshua J. Coon
(DOI: 10.1021/pr900863u).
The data is from experiments in which an S. cerevisiae whole cell lysate was digested with one of five
enzymes (trypsin, LysC, ArgC, AspN, and GluC), in triplicate. The results clearly show that any of these
proteases can be used very effectively with standard proteomics equipment, giving very similar protein
identifications.
New database server added at Rockefeller University (2010/02/05)
Data set of the week: (2010/01/31) Identifying blood biomarkers and physiological processes that distinguish humans with superior performance under psychological stress.
This dataset was transfered to GPMDB via ProteoExchange from PRIDE (Pride accessions 10075-10092).
The data (GPM77710000113-GPM77710000130)
is from a study published in PLoS One by Cooksey AM, Momen N, Stocker R, and Burgess SC
(PLoS One. 2009 Dec 18;4(12):e8371 PubMed).
The results show the plasma proteins that change in response to the Modular Egress Training psychological stress test, given
to a group of naval aviation students. The data was obtained using an LCQ DECA XP Plus and analyzed
using X! Hunter (annotated spectrum library searches).
GPM sites using the new X! Tandem (2010/01/27)
Starting today, the public GPM servers will be using the new release of X! Tanden and X! P3 (2010.01.01.1).
Once live testing is complete, the release code for this new version will be made available.
Features new to 2010.01.01 are improved handling of protein N-terminii and improved
handling of phosphorylated peptides, through the detection of associated neutral losses.
The new parameter set includes the following:
quick acetyl - protein N-terminal modification detection,
stP bias - interpretation of peptide phosphorylation models, and
Data set of the week: (2010/01/24) High quality catalog of proteotypic peptides from human heart
This dataset was transfered to GPMDB from the authors' web site, corresponding to the manuscript of the same name, Kline, KG, et al.,J Proteome Res. 2008 Nov;7(11):5055-61.
PubMed. This data is not currently available on other respositories.
The data consists of 96 LCMS runs
analyzed with a ThermoFinnigan LTQ mass spectrometer. It is a good example of the type of data that can be obtained from cardiac muscle using
multidimensional chromatography directly on tissue lysate.
Data set of the week: (2010/01/17) A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology
This dataset was transfered to GPMDB from TRANCHE, corresponding to the manuscript of the same name, Pagliarini, DJ, et al., Cell 134:112-123
doi:10.1016/j.cell.2008.06.016.
The data consists of 26 individual
data sets, composed of replicates of mitochondrial proteins obtained from a variety of
mouse tissues (cerebellum, cerebrum, brainstem, spinal cord, kidney, liver, heart, skeletal muscle, testis and placenta). It is a good example of high quality proteomics data, obtained using a
Thermo-Finnigan Orbitrap hybrid mass spectrometer.
Data set of the week: (2010/01/10) Comparative analysis of the human and mouse placental transcriptome and proteome
This dataset was transfered to GPMDB from Peptidome via ProteoExchange, from the Peptidome entries PSM1063 (mouse) and
and PSM1064 (human).
The cells in the tissue were separated from extracellular proteins and various subcellular
fractions were analyzed separately. The data was originally published in Cox B, et al., Mol Syst Biol 2009;5:279. PMID: 19536202.
Note: the Peptidome entry misidentifies the mass spectrometry platform as being an "TRAP-FTMS" while it is actually a Thermo-Finnigan LTQ (with no additional hybrid component).
Data set of the week: (2010/01/03) Large-scale phosphorylation analysis of mouse liver
This dataset was transfered to GPMDB from TRANCHE and it is not currently held in any other ProteoExchange database (see data).
It is credited to Villén J, Beausoleil SA, Gerber SA, and Gygi SP, and it is described in Proc Natl Acad Sci U S A. 2007 Jan 30;104(5):1488-93.
This data set is a good example of the quality of phosphorylation data that can be
obtained using SCX separation of a tissue extract, followed by IMAC phosphopeptide
enrichment of each fraction, when using an LTQ-Orbitrap mass spectrometer. The data view that
is obtained from the link above shows all of the detected phosphopeptides, with a peptide
false positive rate of ~ 0.14%, i.e., about 10 times more stringent than the analysis
in the original paper.